A LAMP to see in the molecular darkness

The power of nucleic acid amplification methods—most commonly in the form of polymerase chain reaction (PCR)—is familiar to anyone who has been reading this series. Unsurpassed sensitivity and specificity, with turnaround times on the order of a few hours, have been the features which, along with general applicability of instrumentation, have made PCR-based assays the vehicle for a revolution in medical lab testing.
PCR isn’t perfect, however. It requires the use of instruments (thermocyclers) with highly accurate and reproducible temperature control, capable of rapid shifts between the set points for denaturation, primer annealing, and polymerase extension. It also has to be coupled with a method for product detection—either by opening the reaction tube (risking amplicon contamination of the laboratory) or through application of real-time methods (generally fluorescent in nature, requiring the thermocycler to incorporate complex optical systems for the illumination and observation of reaction tubes in controlled wavelength bands for detection of weak signals, which require complex computer processing to create meaningful data).
Imagine the possible benefits that could accrue if there were an alternative nucleic acid-based technology, with similar speed and sensitivity and good specificity, which could be performed at a single, not-very-critical temperature (suited to, say, a cheap water-bath in a low-resource or otherwise frugal setting), which could be analyzed without additional instrumentation, by eye, for reaction positivity or negativity at reaction end. Such a dream is in fact a reality, and one that is beginning to make the transition from research laboratories to mainstream diagnostics—an assay type known as loop-mediated isothermal amplification (LAMP).
Nothing comes without a price; LAMP’s ability to dispense with the needs for thermocyclers and dedicated real-time or endpoint detection systems comes at the cost of reaction complexity. It has been an aim of this series since its inception to present current molecular methods in a manner which demystifies the technology and applications; while LAMP was considered for inclusion on previous occasions, it has been my humble opinion that within the space constraints of this column it would not be possible to clearly explain in detail how the method functions. Regretfully, I have had no sudden flash of insight to address this, but the increasing frequency with which the laboratorians may encounter this method means this column can avoid it no longer. We shall endeavor, then, to describe the method in a more general form; to supply references and such guidance as is possible for those readers determined to understand the mechanism in its full detail; and to cover aspects of the pros, cons, and types of applications most suitable to the approach. With that disclaimer said, let’s proceed to the method.
How LAMP works
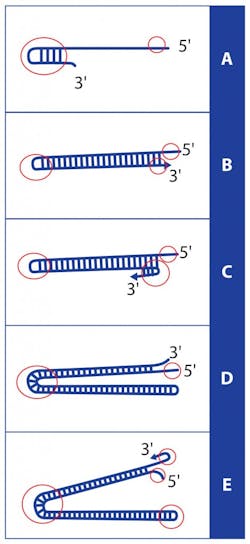
We start from the idea that single-stranded DNA can, when an end of a strand has a sequence which is the reverse complement of a distal sequence on the same strand, fold around to pair the end to the internal sequence in a structure known as a hairpin. (Figure 1A). If the end in question is a 3′ strand end, and the complementarity proceeds right to the 3′ nucleotide, then the hairpin can act as a point of DNA polymerase initiation allowing for extension of the 3′ end back up the hairpin stem, converting the originally single stranded DNA molecule into now an extended double strand (with one end having paired 3′ and 5′ ends, like a conventional DNA double strand, while the other end has a small single stranded loop which is the actual hairpin bend; see Figure 1B). The molecule has in effect, in the presence of nothing more than a DNA polymerase, nucleotides, and a suitable buffer, amplified itself by very nearly a factor of two.
Note however that no complex thermal steps were involved; that specificity arises, much as in PCR, from the complementarity of two DNA regions; and that the strand extension (amplification) could be expected to occur across a relatively wide range of temperatures around the Tm at which the hairpin region is stable, as the hairpin only needs to transiently occur for priming to initiate and an increasingly long and thus stable double-stranded product to ensue. In fact, if this resulting molecule could only somehow “flip” the double-stranded end open in such a way that the 3′ end again could hairpin back along itself (Figure 1C), a strand-displacing polymerase could again initiate and run back the way it just came (and on out to the original 5′ end) in a sort of molecular Jacob’s Ladder (Figure 1D). The double-stranded end then again “flipping” to a hairpin, the polymerase could then continue down what is now a double-length concatamer of the starting single strand molecule (Figure 1E). One could envision that this process continues without end, making a progressively longer and longer molecule in (almost) two-fold steps, until the process stops through reagent depletion. The similarities to classical PCR are obvious, in terms of speed and product yield.
The simplicity of this replication strategy—obviating the need for separate primers, or replication initiation complexes—has not been lost on nature. Those readers who have come across parvovirus B19 (that would be the vast majority of you as unwitting hosts, and a large number of you, professionally dealing with it in the form of erythema infectiosum or Fifth Disease) have met an organism which employs a variation on this strategy as its replication mechanism. (The variation involves a virus-encoded DNA nicking enzyme which cuts and separates the individual viral genomes from the resulting concatamer.) The mechanism, however, also has been adapted by clever molecular biologists, in the LAMP technique.
LAMP then starts by designing a total of four primers (not the two of classical PCR). In the simplest conceptualization of the method, two of these primers each have a section which is complementary to one end of a target DNA section; but each also then has an intentionally “mirrored” section of the target, allowing each primer the capability to form a hairpin. These hairpin primers then delineate the region to be detected through its amplification, if present in a sample. Two additional primers, each nested within the region flanked by the terminal hairpin-forming primers, act as primer initiating points, which cause the “flipping” of the intact double-stranded ends to hairpins needed to continue the replication. Specificity of the reaction then comes in total from the annealing of four custom-designed primers to the target sequence (not the two of classic PCR), and sensitivity from a similar two-fold exponential amplification kinetics.
How, then, do we perform the promised magic of endpoint product detection, just by eye, on the closed reaction tube? The key here is the reaction by-products. PCR (and LAMP) reaction buffers contain relatively high concentrations of Mg2+ ions; these are required to complex with, and in effect “activate” for reaction, the phosphates of the free nucleoside triphosphates (dNTPs) used by the polymerase to extend each nascent DNA strand. Addition of the base and one phosphate from a dNTP to a growing strand cleaves off the remaining two phosphates (known as pyrophosphate or PPi) and the complexed Mg2+ to float free in the reaction. As it happens, though, MgPPi is insoluble and progress of a LAMP reaction can proceed to such an extent that MgPPi becomes clearly visible as a fine, white cloudiness or turbidity in what started as a clear reaction. A variation on this approach is to include a colorometric Mg2+ indicator dye in the reaction, which performs a rather abrupt color change (much like a pH indicator dye) as the Mg2+ concentration in solution decreases through its precipitation.
Obstacles to widespread use
All of this sounds great—so why aren’t we seeing this technique in use a lot more? Well, in the first place, it turns out that LAMP is much more challenging to select primers for than classical PCR. Both the spacing between the four constituent LAMP primers and their relative annealing temperatures (Tm) are much less flexible than in classical PCR, where the thermal cycle can be optimized to Tm of good primers selected for specificity and amplicon parameters. Second, although LAMP relies of four primers for its target specificity, the detection is completely non-specific, and any unwanted side reactions which generate MgPPi can lead to false positive signals. In classical PCR, either a probe-based detection allows for an additional layer of specificity in signal generation—or in binding dye (e.g. SYBR)-based assays, an amplicon Tm check can be used to confirm that a detected signal arises from the intended target. (Note particularly that because of this, LAMP assays should not be incubated beyond the validated time; spurious false positives are likely.) Third, LAMP reactions are not intrinsically well suited to quantitation, while properly configured real-time qPCR can provide quantitative data. Finally, LAMP is at least as sensitive to amplicon contamination and consequent false positive results as is classical PCR. This is a problem of particular importance in low-resource settings where its other advantages of not requiring a complex thermocycler might be most valuable.
So where can we expect to see LAMP assays in use? Their greatest current utility appears to be in a situation such as infectious disease detection in remote low-resource settings, when the pathogen in question happens to have a sequence generally amenable to the designing of a specific LAMP primer set. Particularly when the reagents can be lyophilized down to individual test reactions stable for ambient temperature transport, the ability to merely rehydrate a reaction with sample and incubate in a fixed temperature water-bath followed by direct reading by eye is appealing. Similarly, the method may be increasingly applied to near point-of-care (POC) test environments where simplicity and lack of need for special instrumentation is desirable.
As promised, we shall end our foray into LAMP with both a reference which dedicated readers might wish to follow up on for a more detailed mechanistic description—and a suggestion as to how to approach fully grasping the mechanism. The best reference would be the original paper by Notomi T. et. al. “Loop-mediated isothermal amplification of DNA,” Nucleic Acids Research, 2000;28(12):E63. The associated suggestion would be that the reader be prepared to take a significant amount of paper and pens of multiple colors to sketch out the primer regions, their various inverse complement relationships to one another, and the format of the different intermediates in order to grasp the method. Note that there will actually be both a positive and negative polarity amplification product (not covered in our simplified description, as they don’t conceptually impact anything) and in some versions, there are yet two more short “loop primers” which can increase the amplification yield but again are non-essential in conceptualizing the process as done above or in its actual performance. In conclusion, however, be warned that this exercise in sketching the full reaction is not for the topologically faint of heart!

About the Author

John Brunstein, PhD
is a member of the MLO Editorial Advisory Board. He serves as President and Chief Science Officer for British Columbia-based PathoID, Inc., which provides consulting for development and validation of molecular assays.