Detection of hemoglobinopathies and thalassemias using automated separation systems

Inherited hemoglobin disorders, hemoglobinopathies and thalassemias, largely originated in the tropics, but now are common worldwide due to migration. At least 5.2% of the world population (and more than 7% of pregnant women) carry a hemoglobin variant. It is also estimated that around 1.1% of couples worldwide are at risk for having children with a hemoglobin disorder, and 2.7 per 1,000 conceptions are affected.1
Hemoglobin (Hb) is a polypeptide tetramer; it consists of two pairs of dissimilar globin chains (i.e., α plus β, δ, or γ) and four oxygen-binding heme groups. In healthy adults, ~97% of the total Hb is HbA (α2β2) and < 3.5% is HbA2 (α2δ2). HbF (α2γ2) may be present at trace amounts, if any.
Hemoglobinopathies and thalassemias are two genetically distinct hemoglobin abnormalities. Thalassemias are characterized by a reduced amount of the normal globin chain produced; they result from gene deletion(s) or from mutations. The clinical manifestations of thalassemia can range from mild anemia with microcytosis (β-thalassemia trait) to fatal Hb Barts hydrops fetalis (4 alpha-gene deletions).
The hemoglobinopathies, or Hb variants, are attributable to amino acid substitution(s) in either globin chain. Currently, 1,179 total hemoglobin variants have been characterized.2 Only a few abnormal hemoglobins are common: HbS (the most common worldwide), HbC, HbE, and HbD-Punjab. Most Hb variants are rare or very rare, but, due to the large number of variant hemoglobins, various rare Hb variants are regularly found during routine laboratory diagnostics.3
There are multiple reasons to diagnose Hb variant or thalassemia carriers: 1) to avoid improper treatment of β-thalassemia carriers with iron treatment; 2) to prevent possible complications during anesthesia or heavy physical stress in healthy undiagnosed HbS carriers; 3) to allow genetic counseling for couples at risk;3 4) in a case of diabetes monitoring, to exclude possible Hb variant interference on HbA1c values.
The goal of this article is to provide a brief review of automated methods commonly used for detecting Hb disorders, with an emphasis on capillary separation.
Automated separation methods
Most large laboratories currently use automatic high-throughput methods, such as high-performance liquid chromatography (HPLC) and/or capillary electrophoresis (CE). With virtually 100% sensitivity, these methods easily identify elevated HbA2 in β-thalassemia and common Hb variants.3
High pressure liquid chromatography. On HPLC, hemoglobin samples are injected into a resin column and separated based on charge. Hemoglobin variants elute from the column and are detected at 415 nm, then at 690 nm to correct the baseline. The hemoglobin retention time (from injection until the maximum point of each peak) is calculated and plotted on a chromatogram. Glycosylated fractions and other posttranslational adducts separate from the main peaks, making the chromatogram somewhat challenging to interpret.4
HPLC has been implemented in the clinical laboratory for evaluating hemoglobin abnormalities for more than three decades; an abundance of literature defines the migration of various hemoglobin variants using this technique. HPLC systems provide automation, allow for precise quantitation of HbA2 (with some exceptions), and are effective at detecting common and rare variants. Some rare variants could be missed on HPLC, where they would be detected on CE or other technique, but the opposite is also true.5
Capillary electrophoresis (CE) separation technology. Capillary electrophoresis separates hemoglobin variants by electroosmotic flow and electrophoretic mobility in alkaline buffer (pH 9.4). Multiple samples (two to eight, depending on the instrument used) undergo high-resolution separation concurrently in silica glass capillaries, taking approximately eight minutes to complete the analysis. For Hb variant detection, UV at 415 nm wavelength is used. The detection methodology is similar to one used in HPLC systems. As a result, the methodology is sometimes considered a “hybrid” type of separation technique between classical zone electrophoresis and liquid chromatography.
An electrophoregram consists of 300 consecutive readings and is divided into 15 zones that are either numbered (i.e., Z1) or named according to the common variants (e.g., Z(S) for the zone where HbS migrates). Hemoglobin variants are displayed as peaks, and the zones where the variants belong are automatically marked by the system. All normal hemoglobins (HbA, HbA2 and HbF) are automatically identified. An on-board drop-down library assists with the interpretation of the results.
Significant changes have been made in the latest generation CE systems in order to provide complete automation and improve workflow in the laboratory, while maintaining separation profiles and results identical to those from previous generation instruments. Such systems run two whole-blood programs: 1) Hemoglobin(e) that is used for detection of hemoglobin variants and thalassemias; and 2) HbA1c for measuring HbA1c.
Thalassemia and role of HbA2
No separation technique can be solely relied upon for diagnosing β- or α-thalassemia. DNA analysis is ultimately needed for characterization of the mutations that cause these disorders.3 However, measurement of the HbA2 is essential and commonly used for the routine identification of carriers of β-thalassemia.3,6
In the diagnosis of β-thalassemia trait, it is the proportion of HbA2 relative to any other hemoglobin present that is clinically important, and if the HbA2 variant is present, total HbA2 is diagnostic. In people with β-thalassemia trait, the HbA2 concentration is typically between 4.0% and 6.0%, and is rarely outside the range of 3.5%–7.0% of the total hemoglobin.6
In comparison, α-thalassemia traits will show no specific characteristics on electrophoresis, HPLC, or CE except for a marginal reduction in HbA2 expression. This reduction becomes more prominent when three alpha genes are missing (HbH disease) or are dysfunctional.7 Both HPLC and CE can detect alpha thalassemia in newborns, since various levels of Hb Barts (depending on the number of alpha-gene deletions) will be present in the sample. However, bilirubin can masquerade as Hb Barts on HPLC;8 on CE bilirubin migrates outside of the detection window and does not interfere with the results. HbH and/or Hb Barts can be detected even at concentrations of ~1% on CE. In comparison, 5% concentration of these variants may be necessary in order to discover these variants on HPLC systems.5
A high degree of reproducibility is an essential requirement for HbA2 measurements, due to the very small differences between normal and pathological HbA2 values. Published data for HbA2 quantification for HPLC methods demonstrate low within-run CVs in the range of 0.7%–3.0% and higher between-run CVs ranging from 4.7%-6.0%. Similar studies performed on the latest generation CE systems demonstrated CVs of 2.5% for within- and between-run CVs.9
Beta-thalassemia is not the only reason why the patient may have increased HbA2 concentration. Some Hb variants with a thalassemic phenotype (such as Hb Lepore on HPLC), acquired conditions (megoblastic anemia, hyperthyroidism, hypertrophic osteoarthropathy), and even antiretroviral therapy in patients with HIV can lead to increased HbA2. Iron deficiency anemia, a very common disorder, is one of the main causes of low HbA2 values.6
When interpreting Hb results, CBC results always should be reviewed. Thalassemia carriers are anemic with microcytic and hypochromic parameters that mimic those in iron deficiency, but RBC is typically elevated in thalassemia. Serum ferritin and/or zinc protoporphyrin levels should be reviewed to exclude possible iron deficiency.3
Effect of hemoglobin variants on HbA2 quantification
Common Hb variants can interfere with HbA2 quantification on HPLC. Glycated HbS fractions coelute with HbA2 on HPLC; as a result, the HbA2 fraction is overestimated.4,10 On rare occasions, this may lead to a misinterpretation of the results (HbS/ β-thalassemia) by operators.3 In a non-transfused Hb S homozygote or a compound HbS/β0 thalassemia, HbA is not present, but peak(s) due to HbS adducts often elute in a position similar to HbA and get labeled as HbA, hence changing the apparent genotype from homozygous HbS to HbS/β+ thalassemia.6
HbD elutes close to HbA2; it generally leads to an underestimated HbA2 value on HPLC. Hb Lepore and HbE co-elute with HbA2 on HPLC. If HbA2 peak is around 12%-16% on HPLC, Hb Lepore should be suspected since the maximum value of the HbA2 fraction in beta-thalassemia trait is 8%-9%. HbE can be putatively identified if the peak size is 20% to 25%.3
On CE systems, common hemoglobin variants (HbS, HbC, HbD-Punjab, and HbE) migrate in different zones. Common Hb variants separate from HbA2. Glycated Hb fractions or other adducts do not separate from main fraction, do not interfere with HbA2 quantification, and make interpretation of patterns easier.4,10,11 When using CE for separation of HbE and HbA2 in heterozygous individuals, slightly increased HbA2 values (3.5% ± 0.4%) were observed confirming β+-thalassemia phenotype of HbE.12
Delta chain variants are easily detectable using CE and HPLC. Stable δ-chain variants may appear as a second HbA2 fraction migrating/eluting either faster or slower than normal HbA2 and, typically, they are expressed at an approximately equal level as HbA2. HbA2‘ is the most common delta-chain variant, and is one of the most common hemoglobin variants, particularly in the African-American population.5 If delta chain variant is present in the sample, its value must be added to HbA2 in order to accurately report total HbA2.6
CE is very efficient in detecting Hb Constant Spring (Hb CS) that contains a point mutation extending α-globin chain by another 31 amino acids. The variant is unstable and constitutes 1% to 2% of the total Hb in heterozygotes. Non-deletional HbH disease, the result of interaction of the Hb CS with deletional alpha-
thalasesemia, is typically more severe than deletional Hb disease. In Hb CS trait, red blood cell (RBC) parameters are within the normal range, and screening is impossible using RBC indices alone. As a result, other screening methods are required. In a recent study, 100% of patients with known Hb CS trait were detected using CE system, while 76.2% (16 out of 21) were detected using HPLC.13
Diabetes mellitus and hemoglobin variants
It is estimated that 25.8 million children and adults in the United States (8.3% of the population) have diabetes; 79 million people have prediabetes. The International Diabetes Federation (IDF) has reported the prevalence of the disease in 2011 as 366 million worldwide. Countries with the highest incidence of diabetes also tend to have high incidence of Hb variants in the population.14
Currently, the American Diabetes Association recommends using HbA1c as a marker to diagnose diabetes using an NGSP-certified method and a cutoff of HbA1c of ≥ 6.5%. HbA1c, the major form of glycated hemoglobin, is the result of the non-enzymatic glycation of the N-terminal valine residues of the β chain of HbA.15
Inaccurate HbA1c values are obtained when Hb variant, or its glycated derivative, cannot be separated from HbA or HbA1c. HbF, Hb Graz, and Hb Raleigh have been shown to cause decreased HbA1c values by immunoassay; other Hb variants with alterations in the first 4 to10 N-terminal amino acids could produce similar results. Boronate affinity methods are thought to be free of Hb variant interference, but Hb Himeji can increase HbA1c value, and even common Hb variants can affect performance of some boronate systems. The effect of Hb variants on various HPLC systems is diverse, system-dependent, and well documented.16
Correct interpretation of HbA1c measurements depends on normal erythrocyte lifespan. In individuals with sickle cell, HbC, or HbD disease it is recommended that tests other than HbA1c be used for the determination of glycemic control (e.g., glycated serum albumin), since the lifespan of red blood cells is altered. If heterozygous carriers show normal erythrocyte survival, HbA1c can be used as long as the hemoglobin variant does not interfere with the assay method, nor with glucose binding to hemoglobin.17 There is some evidence to suggest that Hb variants may affect RBC lifespan even in Hb trait patients who are asymptomatic.14 Thalassemia also can affect life span of RBC, resulting in a markedly low HbA1c value.18
Despite the advances in diagnostics, the status of hemoglobinopathies and/or thalassemias in many patients is unknown. HbA1c techniques are optimized for speed in HbA1c testing; most provide no or limited information about Hb makeup in the sample. With very few exceptions, no HbA2 values are displayed, rendering detection of thalassemia impossible. However, ability to detect hemoglobinopathies in the sample is clearly desirable. especially, in populations with high ethnic diversity.
The latest assay, based on CE separation, measures HbA1c using the IFCC calculation formula without interferences from labile HbA1c, HbF, acetylated or carbamylated HbA, and/or common Hb variants.15,19 It processes 38 whole blood samples per hour, and up to eight samples can be run simultaneously. HbA1c, minor glycated Hb, HbA0, HbA2, and HbF peaks are separated and automatically identified by the system. The software flags the presence of Hb variant by displaying an “Atypical Profile” message on the result screen and color-coding the electrophoregram. Common Hb variants have specific migration positions on the pattern,15 although no migration zones are displayed. Delta- and alpha-chain variants can be easily visualized using HbA1c program; they present with patterns similar to those observed on the Hemoglobin(e) program (Figure 1).
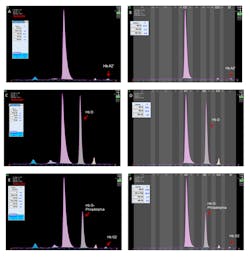
Figure 1. A,B. HbA2’ (delta chain variant) in the sample run on HbA1c and Hemoglobin(e) program, respectively. C,D. HbA/D sample run on HbA1c and Hemoglobin(e) programs. E, F. HbA/G-Philadelphia (alpha-chain variant) run on HbA1c and Hemoglobin(e) program. (Click on the image to see a larger version)
A complete separation of the HbA2 quantification between non-carriers and carriers of the β-thalassemia trait has been demonstrated using this latest HbA1c method.19 The software displays an alert when HbA2 value is higher than 3%.
Currently, the HbA1c program is FDA-cleared for separation and quantification of HbA1c only. If Hb disorders are visually seen on the electrophoregram, they should be confirmed by the Hemoglobin(e) program on the same analyzer.
Once a hemoglobinpathy is detected, a different technique for confirmation of abnormal hemoglobin results should be used. By combining CE and HPLC, one can reduce the number of unusual hemoglobin variants that can be confused with normal hemoglobins or common Hb variants.
Aigars Brants, PhD, serves as Scientific Affairs Manager for Sebia, Inc., providers of the CAPILLARYS 2 Flex Piercing CE system. www.sebia-usa.com
References
- World Health Organization. Global epidemiology of haemoglobin disorders and derived service indicators. http://www.who.int/bulletin/volumes/86/6/06-036673/en/. Accessed December 3, 2013.
- HbVar: A database of human hemoglobin variants and thalassemias.
http://globin.cse.psu.edu/cgi-bin/hbvar/counter. Accessed December 3, 2013. - Giordano, PC. Strategies for basic laboratory diagnostics of the hemoglobinopathies in multi-ethnic societies: interpretation of results and pitfalls. Int J Lab Hemat. 2013;35(5):465-479.
- Keren DF, Hedstrom D, Gulbranson R, Ou C, Bak R. Comparison of Sebia capillarys & capillary electrophoresis with the Primus high-pressure liquid chromatography in the evaluation of hemoglobinopathies. Am J Clin Pathol. 2008;130:824-831.
- Greene DN, Pyle AL, Chang JS, Hoke C, Lorey T. Comparison of Sebia capillarys flex capillary electrophoresis with the BioRad Variant II high pressure liquid chromatography in the evaluation of hemoglobinopathies. Clin Chim Acta. 2012;413(15-16):1232-1238.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol. 2012;34(1):1-13.
- Galanello R, Cao A. Gene test review. Alpha-thalassemia. Genet Med. 2011;13(2):83-88.
- Howanitz PJ, Kozarski TB, Howanitz JH, Chauhan YS. Spurious hemoglobin Barts caused by bilirubin: a common interference mimicking an uncommon hemoglobinopathy. Am J Clin Pathol. 2006;125(4):608-614.
- Agouti I, Merono F, Bonello-Palot N, Badens C. Analytical evaluation of the Capillarys 2 Flex Piercing for routine haemoglobinopathies diagnosis. Int J Lab Hemato. 2013;35(2):217-221.
- van Delft P, Lenters E, Bakker-Verweij M, et al. Evaluating five dedicated automatic devices for haemoglobinopathy diagnostics in multi-ethnic populations. Int J Lab Hematol. 2009;31:484–495.
- Mais DD, Gulbranson RD, Keren DF. The range of hemoglobin A2 in hemoglobin E heterozygotes as determined by capillary electrophoresis. Am J Clin Pathol. 2009;132:34-38.
- Winichagoon P, Svasti S, Munkongdee T, et al. Rapid diagnosis of thalassemias and other hemoglobinopathies by capillary electrophoresis system. Transl Res. 2008;152(4):178-184.
- Liao C, Zhou JY, Xie XM, Li J, Li R, Li DZ. Detection of Hb Constant Spring by a capillary electrophoresis method. Hemoglobin. 2010;34(2):175-178.
- Rhea JM, Roberts-Wilson TK, Molinaro RJ. Impact of hemoglobin variants on Hb A1c interpretation: Do we assume too much? MLO. 2012;44(6):8-14.
- Jaisson S, Leroy N, Meurice J, Guillard E, Gillery P. First evaluation of Capillarys 2 Flex Piercing (Sebia) as a new analyzer for HbA1c assay by capillary electrophoresis. Clin Chem Lab Med. 2012;50(10):1769-1775.
- Bry L, Chen PC, Sacks DB. Effects of hemoglobin variants and chemically modified derivatives on assays for glycohemoglobin. Clin Chem. 2001;47(2):153-163.
- Hinzmann R, Schlaeger C, Tran CT. What do we need beyond hemoglobin A1c to get the complete picture of glycemia in people with diabetes? Int J Med Sci. 2012;9(8):665-681.
- Bry L, Chen PC, Sacks DB. Effects of hemoglobin variants and chemically modified derivatives on assays for glycohemoglobin. Clin Chem. 2001;47(2):153-163.
- Urrechaga E. High-resolution HbA(1c) separation and hemoglobinopathy detection with capillary electrophoresis. Am J Clin Pathol. 2012;138(3):448-456.
