Back to Basics: RNA molecular diagnostics

While much of molecular diagnostics (MDx) deals with deoxyribonucleic acid (DNA) as its target material, this month’s “Back to Basics” Primer topic will pause to consider “the other nucleic acid,” ribonucleic acid (RNA): why it can be the target of choice or even necessity for some MDx methods, and what some of the general challenges are in handling RNA in the lab.
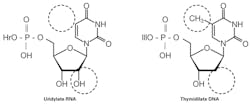
First, a quick refresher on the chemical difference between RNA and DNA. In either case, a nucleotide—the building block for a nucleic acid strand—consists of a nitrogenous base, a sugar, and phosphate(s). The critical difference is in the sugar, where RNA has a 2’ –OH group not found in DNA (Figure 1). It is this missing piece that leads to the “D” in DNA’s name—Deoxyribonucleic acid. There’s also a difference in that, while the bases adenine, guanine, and cytosine are common to both RNA and DNA, the DNA base thymine is substituted by uracil in RNA. Base pairing rules, however, are the same, and this base difference isn’t generally a conceptually significant concept.
On a macrostructural level, a more significant difference between DNA and RNA is that RNA is generally a single-stranded macromolecule, while DNA is double-stranded. This allows much more strand flexibility than DNA has, and lRNA will take advantage of this to frequently fold back on itself and pair self-complementary regions in various stem-loop configurations. These structures can have specific functional purposes in an RNA molecule, independent of any informational content carried purely on a nucleotide-sequence basis.
Why do RNA MDx?
If all living organisms use DNA as their genetic storage material, it’s tempting to ask why would we be interested in looking at RNA at all? A first answer is that it’s somewhat disingenuous to only consider strictly living organisms as being clinically significant. Viruses do not count as living, yet it is an understatement to say that many are of clinical importance—and many viruses are not only RN-based but never go through a DNA stage in their replication cycle. A first key reason for RNA-based MDx methods, then, is to detect these types of pathogens.
If we stay within the infectious diseases (ID) context for the moment, there can also be benefits to choosing RNA as our target material even in living, DNA-based organisms. One such reason is that by nature, with a single DNA gene copy being potentially transcribed hundreds or even thousands of times, we may be able to effectively increase our detection thresholds by an equivalent amount; that is, testing for the expressed RNA of a characteristic gene marker may allow for the use of smaller samples and/or the detection of an organism at much lower levels than direct DNA-based MDx.
A second reason is an extension of this, in which we consider that RNA is a marker for not just gene (and thus organism) presence but more specifically for its biological activity as evidenced by ongoing gene expression. A shortcoming of DNA-based MDx in ID can be that it doesn’t differentiate between viable organisms and leftover unviable remnants; because of RNA’s much lower stability (discussed further below), RNA-based assays can be a more direct indication of ongoing infection than their DNA counterparts. This data may even directly relate to probable pathogenicity, such as in the example of HPV; evidence suggests that even among infections with known high-risk genotypes, active expression of the E6 and E7 genes is a more likely indicator of progression to significant disease than is mere presence of intact but transcriptionally inactive virus.
This concept of examining RNA as a window into transcriptional activity is also how RNA-based diagnostics comes into use outside of the ID field. Genetic diseases arising through dysregulation (over- or under-expression) of a gene aren’t distinguishable by traditional DNA-based MDx, particularly in examples where no variation in gene copy has occurred but may be readily distinguished by RNA-based quantitative methods such as real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) or RNA gene expression arrays. In addition to permanent genetic changes such as promoter activation or inactivation, these methods can also be employed to capture information on transient variations in gene expression related to pathologic processes such as immunological responses.
Why not do only RNA MDx?
We have established above that in some cases RNA is the only possible MDx target, while in others it offers advantages in sensitivity or insight to gene/organism activity over DNA targets. With all of these features in its favor, our question now is not “Why do RNA MDx?”—but rather, “Why do DNA MDx when we could do RNA?”
Much of the answer to that comes back to the existence of that 2’ –OH group in RNA and its impacts on molecular stability. In the simplest summary, this “extra” hydroxyl group relative to DNA makes RNA much less chemically stable than DNA by virtue of its being prone to hydrolytic cleavage events along the sugar-phosphate backbone. This intrinsic lower chemical stability of RNA is vastly accentuated by the pervasive prevalence of RNases—enzymes dedicated to the degradation of RNA molecules. In fact, from a biological perspective this enforced instability of RNA is, as people in the software world would say, “a feature, not a bug!”
Consider that a cell reacting to local environmental cues wants to alter its activities in response, and in many cases does so by altering (up- or down-regulating) transcription of one or more relevant genes. For this sort of environmental response to be dynamic, a rapid turnover of RNA transcripts is essential so that increases or decreases in RNA transcription actually relate within a reasonable time period to increases or decreases of the steady-state level of the transcript (and thus either its direct function or its capacity to drive protein translation, for a coding mRNA). RNA, then, is a great template when you can get it intact, but ensuring that this is done is quite a lot more challenging than in obtaining intact DNA.
In order to be able to handle RNA in the laboratory, it is important to take steps to mitigate these stability issues. Intrinsic chemical instability is addressed through use of carefully pH-adjusted sample buffers (readily done, and usually done for DNA as well, although it’s not quite as essential). Where sample handling and storage needs begin to diverge from those of DNA are in long-term storage, where -80°C is recommended for RNA while -20°C is often sufficient for DNA. (Lest anyone panic, short-term storage of RNA samples at -20°C is generally fine).
More important are steps taken to inactivate RNases that are lurking everywhere. This involves steps such as ensuring all reagents are RNase activity-free but, as RNase enzymes are remarkably robust, simple autoclaving of reagents will not suffice, and this is generally done in-house through use of specific chemical inactivators and/or inhibitors of RNases. Many reagents can also be purchased (at a premium) as intended for RNA use and arrive so prepared. Use of personal protective equipment (PPE), already essential for protection of staff from materials being handled, gains an additional level of urgency in handling of RNA samples, where it must also protect the specimen from the staff. Similarly, many labs will enact practices such as keeping designated pipettor sets for RNA work. With these mitigations in place to ensure template stability, we can now consider some of the mechanistic changes needed in performing MDx work on RNA templates.
MDx on RNA templates
To a first approximation, any PCR-based assay intended for use on a specific DNA target can be modified to work on its RNA transcript by addition of a single preliminary stage to the existing PCR protocol. This first stage utilizes a retrovirally derived enzyme known as a reverse transcriptase (RT), which as the name suggests takes an RNA transcript and converts it back to a single conjugate DNA strand. This reverse transcription activity initiates at a section of the RNA hybridized to a primer, allowing, for example, one of the pre-existing PCR primers to perform this function and direct reverse transcriptase activity to region of interest.
While it is possible to perform a standalone reverse transcription reaction and then manually use the products of this as a template for a second PCR reaction, the additional handling steps and potential for contamination inherent in such a two-step process make it undesirable in a clinical lab setting. While it is less likely to be ideally optimized for maximal sensitivity, a combined reaction mixture containing both PCR and RT reagents is more practical. In this context, the most common approach involves an initial lower-temperature reaction phase generally around 42°C for 15 to 30 minutes that provides the RT step, followed by a high-temperature step for a few minutes to deactivate the RT enzyme and trigger function of a heat-activated PCR polymerase, followed by regular PCR thermocycling. Within this outline, PCR variations such as real-time techniques, touchdown or thermal gradient steps, and the like can all be performed as they would be on a plain DNA template.
In some contexts, additional considerations of the underlying biology remain useful—particularly if the goal is to specifically detect RNA such as an actively produced transcript distinct from its DNA gene of origin. From the description of an RT-PCR assay above, it’s possible to imagine how either an RNA or DNA copy of the region flanked by the PCR primers could be amplified. If it’s critical to know that a resulting positive signal (amplicon) can only be RNA-derived, then one option is pre-treatment of the RT step template material with DNase enzymes. If this approach is taken, it is important that the DNase be of high purity (no RNase contaminant) and that it be used under conditions which do not lend themselves to side RNA-degrading activity. In the case where the target RNA is from a eukaryotic organism, a simpler solution may sometimes be available by selecting PCR primers from different exons, such that they are close together in an RNA transcript and readily amplified but separated by large intron(s) on the genomic DNA. The genomic DNA target will then fail to amplify, and only spliced RNA will yield product.
In conclusion, I hope the reader will appreciate how RNA can be a valuable or even essential target for MDx applications, but its use requires additional steps and complexities. This ensures that it won’t supplant DNA as target of choice in all applications.
One final bit of interesting related information: recall from above that one base in RNA (uracil) is functionally replaced by another in DNA (thymine). The difference between these bases is a single methyl (-CH3) group present on thymine but absent in uracil. This methyl group is not significant from a chemical reactivity standpoint, so what is its biological use? The answer to this comes in realizing that uracil is also structurally similar to the other DNA pyridine, cytosine, having an exocyclic oxygen where cytosine has an amine (-NH2). It turns out that a very common form of DNA mutation can arise due to spontaneous hydrolytic deamination of cytosine, where its amine is displaced by an oxygen—in fact turning it into a uracil. If this were not detected, during the next DNA replication cycle the base added to the nascent strand at this point would be an adenine (A-U base pair, equivalent to an A-T base pair) instead of the correct guanine (originally pre-mutation paired to the cytosine). By having thymine residues carry a methy group “flag,” as it were, DNA repair machinery in the cell can detect any uracils present as being “wrong” and trigger a DNA repair mechanism which excises the uracil and replaces it with a correct cytosine before the mutation can be fixed in the genome.
John Brunstein, PhD, is a member of the MLO Editorial Advisory Board. He serves as President and Chief Science Officer for British Columbia-based PathoID, Inc., which provides consulting for development and validation of molecular assays.
About the Author

John Brunstein, PhD
is a member of the MLO Editorial Advisory Board. He serves as President and Chief Science Officer for British Columbia-based PathoID, Inc., which provides consulting for development and validation of molecular assays.