
In last month’s installment, we covered some of the ways in which a ligation step can be added into (or before) a PCR-based methodology to improve one or more aspects of the assay. While the topic of this month’s article—padlock probes—also fits this description, it’s unusual enough in its design and assay features to deserve its own column.
PCR reactions are, by and large, rather individualistic and temperamental things. Optimization of any reaction is a balance between sensitivity and specificity, chosen by tailoring reaction chemistry (components and concentrations) and conditions (times and temperatures of the thermocycling profile). In the context of a simplex (single-target) reaction, all of these can be optimized at will, but if the goal is a multiplex reaction, things get a lot more complicated.
Selecting all primer sets to have similar melting temperatures is probably the most crucial step, and to do so while ensuring uniqueness and thus specificity of primer binding sites generally leads to a range of amplicon sizes and differential amplification efficiencies of the separate amplicons in the mixture. While this can be compensated for to some extent by things such as differential primer concentrations, in the end a multiplex traditional PCR must be a sum of many compromises to reach acceptable (but almost certainly suboptimal) performance of each of the component singleplex reactions. It’s just a natural consequence of the need for different primer sets, amplicon sizes, and dissimilar probe or hybridization capture regions within each singleplex.
Imagine now if there were a method that could multiplex large numbers of singleplex targets, but do it in such a way that a single common primer set could amplify every target (no more compromising on annealing temperatures); all targets could be the same length (no more compromising on extension times); and a real-time probe or hybridization capture tag sequence could be engineered in, assuring both that each possible amplicon would be readily distinguishable and that the probe or hybridization capture tags would all have very similar annealing behavior. It’s as if you’re promised a magical beast which does away with many of the classical challenges of multiplexing. Such an assay method does exist in the form of padlock probes. As always, though, there are costs; read on to discover the method, and what those costs are.
Padlock probe basics
The basic concept of a padlock probe is outlined in Figure 1. Essentially, it consists of a single-stranded synthetic DNA probe designed against each target of interest (a). The 5′ and 3′ termini of this probe are target-specific; they’re designed to be complementary to two immediately adjacent sequences of the target nucleic acid, labeled here as R1 and R2. If the padlock probe is mixed with target nucleic acids containing these sequence complements, thermally denatured and reannealed, it can hybridize down to its target at both ends, effectively circularizing the probe and placing its 3′ and 5′ ends immediately next to each other, but with a nick—that is, missing the phosphodiester bond; see Figure 1b. If a DNA ligase is present—preferably, one from a thermostable organism so it could have been added during reaction setup, prior to thermal denaturation—it can now act at this nick, converting a previously linear padlock probe into a covalently closed circular molecule; see Figure 1c. In some versions of this type of assay, we will actually run a few cycles of denaturation / annealing / wait for possible ligation in an effort to drive formation of multiple copies of this circular product.
Of course, this doesn’t occur to every padlock probe we added into the reaction, but only to a small fraction. That’s fine, and to get rid of uncircularized probes we now add in single strand-specific DNA exonucleases. These “chew up” linear single-stranded DNA but leave any circular molecules intact. Now all we have to do is detect these.
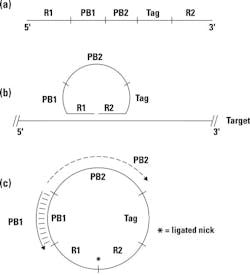
That’s where we switch back to classical PCR—eithker in a real-time format or upstream of a hybridization capture-based detection (that is, a microarray). If you look back at Figure 1, you’ll note that on the padlock probe, internal to the target-specific R1 and R2 ends, are a pair of primer binding sites PB1 and PB2. The key here is that these work with a pair of primers; let’s call them PB1′ (complement to PB1) and PB2 (identical in sequence to PB2; see Figure 1c). If we conduct a standard PCR reaction with these primers and have any circularized padlock probe present, consider what happens. PB1’ binds to the circularized probe and gets extended across the now-ligated R1/R2 junction, and out across PB2, creating a complement to PB2, and thus a binding site for the PB2 primer. Consider that if the padlock probe hadn’t circularized, the nascent strand growing from PB1′ would have run out of template and stopped at the R1/R2 junction, thus never reaching PB2. Only a circularized template allows for PB1’ to extend across the PB2 site, creating what is now a “normal” linear PCR template for the PB1′ / PB2 primer pair. PCR amplification proceeds normally from there on.
Further observations
There are two things to observe at this juncture. First, note that any number of padlock probes could be used simultaneously with differing R1/R2 (target-specific) regions, but sharing a single PB1′/PB2 amplifying primer pair. That’s where our promise above of using a single common primer set to amplify disparate targets comes from—so we only have to optimize PCR for this single PB1′/PB2 primer set (and note as well that we can design these sequences at will to have convenient thermal characteristics, lack of secondary structures, and other desirable characteristics; they aren’t constrained to be part of any assay target sequence).
A second possible observation: what if instead of classical PCR, we used just the PB1′ primer, a polymerase, and did an isothermal amplification? If you’re guessing this would leave the polymerase racing around and around the closed padlock probe template and creating an ever-lengthening nascent strand consisting of concatemer linear copies of the circular sequence, you’re right. Known as rolling circle replication or RCR, that’s something we could do here as well. It would not be quite as specific as direct PCR (needing both primers PB1′ and PB2 to match) and wouldn’t yield as much signal amplification as classical PCR, but it would effectively tether the growing product to the point of amplification. RCR-based padlock probe detection is therefore sometimes used for in situ molecular detection, where we want to know localization of targets to specific cell types such as in a tissue thin section. For the sake of brevity though, we’ll stick to just the classical PCR-based detection of our padlock probe for the remainder of this article.
So that then brings us to the PCR detection part. If we assume we have probed a sample with several different padlock probes (remember, each type with unique R1/R2 regions but shared PB1/PB2), and all of identical or close to identical total length), how do we tell them apart after our PCR has amplified up any probes which circularized? The answer here is given in Figure 1 as well. Note that we’ve incorporated a unique tag sequence in each padlock probe. Much like the PB1/PB2 sequences, these are arbitrary and up to the whims of the designer, meaning they can be chosen to provide highly selective hybridization capture tags (that is, complements to a series of spots in a premade microarray) or binding sites for real-time PCR probes suitable for our choice of probe-based real-time chemistry. Again, we can pre-design a whole set of such probe sequences optimized for similar hybridization behavior with minimal cross-hybridization, bothersome secondary structures, or other problems. The answer to our method of detection then can be either of real-time PCR (probe based against the tag) or microarray hybridization (against the tag). Obviously, if we’re going to multiplex more than four to six targets simultaneously—the practical limit imposed by spectral resolution issues in real-time PCR—then array-based detection is preferable. Keep in mind, however, that we needn’t make a new array each time we want to change a target of our multiplex assay; we only need to change the target-specific R1 and R2 regions of the padlock probe which carries a predetermined array tag. Of course the same argument applies if we did use real-time detection, too.
This final point—that we could premake a microarray, or set of real-time probes, and mix and match these against targets at will without redesign of the amplification primers or detection probes or array—is the icing on the cake, as it were, to our laundry list of desirable features which padlock probes have as a methodology over more traditional multiplex PCRs.
Considering the costs
The costs, then? These occur in the form of higher reaction complexity in terms of what has to go together and work in the reaction tube, not just PCR reagents but also a ligase and exonucleases. Since we don’t want the ligase or the exonucleases working later on in the reaction, these have to be effectively inactivated, usually by an extended, high temperature step, after their point of action. The hybridization kinetics of a long padlock probe to target is not generally as fast or good as short, classical PCR primers, so lower limits of detection by this method may lag behind those of more direct PCR. Finally, synthesis of padlock probes, on the order of 100 bp in size, is more expensive and lower yield than synthesis of shorter, traditional PCR primer-sized molecules (However, compared to a few years ago, costs have decreased and yields increased significantly for longer oligonucleotides such as this, making that less of a hurdle than it used to be.)
Where is the laboratorian today likely to come across this type of assay? The most common application at present is probably in the detection of single nucleotide polymorphisms (SNPs). By placing the SNP of interest directly under either side of the R1-R2 junction, a mismatch to padlock probe will block ligation, or, conversely, a match will allow ligation and selective signal generation only when a perfect match occurs. Small insertions or deletions under the R1/R2 target area will also block effective circularization. Beyond such genotyping applications, other uses in the literature have included multiplex pathogen detection assays.
John Brunstein, PhD, is a member of the MLO Editorial Advisory Board. He serves as President and Chief Science Officer for British Columbia-based PathoID, Inc., which provides consulting for development and validation of molecular assays.
About the Author

John Brunstein, PhD
is a member of the MLO Editorial Advisory Board. He serves as President and Chief Science Officer for British Columbia-based PathoID, Inc., which provides consulting for development and validation of molecular assays.