Oncogene panels: a window into the individuality of cancers

Cancer has been a scourge of humanity for as long as we know, with written descriptions extending back at least as far as the Ebers and Smith papyri, estimated to have been written around 1600 B.C. and postulated to be derived from earlier sources from circa 2500 B.C. As such, it has been subjected to intensive study, perhaps more so than any other single class of ailment. The pace of progress in battling cancers has steadily accelerated, and a relatively new tool—the oncogene panel—is beginning to prove its value. In this month’s Primer we’ll examine briefly what these MDx tools do, and how they are both changing our understanding of cancer and, in some instances, allowing for effective application of what might otherwise have been unlikely treatment options.
On the surface, cancers are misleadingly simple pathologies. Cells making up the various organs and systems of the body can replicate, and normally only do so when required for development or repair; replication ceases when the cells making up an organ system reach their required size and functions. Simplistically, cancer occurs when replication of a cell becomes uncontrolled and a single cell divides repeatedly to become a tumor, ignoring and overrunning surrounding tissues. This physical disruption of well-behaved cells by tumor tissue leads to disruptions in proper organ function and all of the downstream consequences.
Traditionally a major tool in classifying and directing treatment of cancers has been through identification of the underlying cell type which has become dysregulated. Cancers sharing an origin type such as lymphoma, lung cancer, or colon cancer might reasonably be expected to share commonalities in pathology, disease progression, and best treatment strategies. In reality, however, it is observed that cancers sharing overt similarities in terms of cell type, appearance, and progression stage may respond quite differently to the same drug treatment. We are beginning to appreciate that for some cancer types that have been known under a single name, individual cases can be just that—individual, with regard to the root defects and thus best treatment strategies.
To understand this a bit better and the role of oncogene panels, we should go back to the basic concepts of cell division and its regulation. There are large numbers of highly complex multistep biochemical pathways which either act to turn on cell division or to shut it off. Individual cells are controlled by several such pathways simultaneously, and for progression to cancer generally several of these pathways have to be disrupted in a coordinated fashion. This adds complexity to the underlying cause, but also suggests that effective treatment might be possible through blocking any one of the dysregulated paths. The questions, then, are: “Which path?” and “Where can we block it?” These are the questions that oncogene panels seek to directly answer.
Pathways to malignancy
Let’s consider a hypothetical pathway relating to cell division, as sketched out in Figure 1. This consists of an extracellular signal “ligand”(1); a trans-membrane receptor spanning across the membrane of the cancer cell, with extracellular receptor domain (2); an intracellular effector domain (3); an intracellular secondary messenger (4); and a transcription factor (6) with its inhibitory partner (5).
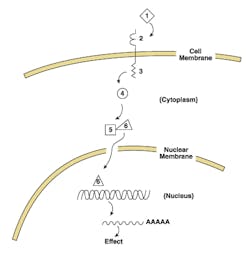
If this represents a pro-replication signal pathway, then it would function effectively as follows: a positive signal (such as a growth hormone (1), binds (2), causing an activating conformational change in (3); this enzymatically transiently modifies (4), which in its modified state causes inhibitory subunit (5) to release (6), which now translocates to the nucleus, binds specific signal sequences in the DNA upstream of a pro-replication gene, and upregulates or induces transcription of the product RNA, which then is translated to an active protein which functionally proceeds to drive a cell division cycle (providing other required pro-replication factors are present, and anti-replication factors are absent—thus the above statement on need for multiple coordinated errors before cancer occurs). This whole pro-division pathway acts for a finite time, as the ligand at (2) is released or internalized and degraded, the modification of (4) decays off, and the RNA transcript and final protein product both undergo normal turnover processes. Note that we could just as easily postulate this model for an anti-replication pathway, if we called ligand (1) a negative signal such as contact inhibition, and the RNA codes for a replication inhibitory protein; for sake of argument, though, we’ll stick to a pro-replicative model for now.
With this pathway model in mind, let’s ask ourselves what different changes could occur to be cancer-causing (pro-oncogenic). Some immediate possibilities could include:
- Excess or inappropriate expression of ligand (1);
- Alteration of the transmembrane receptor, either in domain (2) or (3), such that it behaves as if ligand is bound even when it’s not;
- Alteration of secondary messenger (4) so it’s constitutively active, thus constantly relaying a signal from (3) that’s not really there; or
- Alteration of (5) such that it loses ability to sequester and inactivate (6).
These are hardly exhaustive or exclusive, and the reader is invited to try to postulate some other possible pro-oncogenic changes to our model path. For instance, consider alteration of the DNA creating a binding site for (6) upstream of a pro-replication gene where it shouldn’t be, such that a signal intended for something unrelated to replication is now “understood” by the cell as a signal for replication. If that sounds far-fetched, it isn’t; essentially that’s the basis of Burkitt’s lymphoma, where the promoter DNA for IgG heavy chain production gets fused in front of a pro-replication gene c-Myc; signals for the cell, which are intended to cause it to make antibodies, now make it go into a cell-division cycle.
Clinical implications
The point of this hypothetical pathway and exercise in imagining all the ways in which it can become dysregulated is to begin to appreciate that two cells of the same type, both of which have become cancerous through inappropriate activation of the same pathway, may have done so through very different underlying changes. Both will present, on the surface (or perhaps we should say, in thin sections below the surface, as seen through the pathologist’s microscope) as very similar, or even identical. Likely both will respond similarly to crude chemotherapies, such as those which generically block DNA replication; however, those chemotherapies are also, for obvious reasons, the ones with the worst side effects. As our knowledge of the mechanisms of cancer have increased, so too have the clinician’s options for highly targeted drug therapies which can block or interfere very specifically with steps on the pathway. For oncogenic pathways similar to our model, drugs exist which can bind and inactivate (2) from binding excess or ectopic ligand (1); others block the activation of secondary messenger (4), or interfere with the binding of (6) to its target DNA sequence.
In the right setting, these drugs can be highly effective. For instance, some types of lung cancer originate through dysregulation of a pathway very similar to our model, through overexpression of ligand—and highly specific drugs have been developed which bind and mask the receptor extracellular domain (2) to stop it from responding to the ligand. These drugs, while expensive, are highly effective, in this case in halting further progression. However, in some cases which present with identical appearance, the defect occurs in (4), (5), or (6)—in the parlance of signal transduction biology, “downstream” from the receptor and point of drug action. In these cases administration of the drug is ineffective and possibly harmful, as valuable time is lost before observing that it’s ineffective.
The key to using these pathway and step-selective drugs in an effective manner is therefore to know, on a highly individual basis, what the exact set of genetic changes in the cancer cell is, so that a drug with appropriate point of action can be selected—something downstream of at least one of the critical mutations. (Remember, usually multiple coordinated mutations must be active to result in full-on cancer; so interfering with even one of these may be enough to stop progression.) Thanks to years of research and the Human Genome Project, cancer researchers now know of literally hundreds of genes whose products play known roles in the multiple pathways promoting or supressing cell division. They also have detailed libraries of mutations in these genes, known to lead to unwanted activation or inactivation of the gene product.
Oncogene panels
An oncogene panel is a targeted next-generation sequencing (NGS) approach, which can take a patient sample (from tumor tissue, not unaffected tissue) and simultaneously sequence all of these hundreds of understood genes on cell division control pathways. The exact platform and sequencing method is unimportant; from a technical standpoint, however, it is worth noting that the amount of sequence data needed per patient is relatively small on an NGS scale, allowing for multiplexing of many patient samples in a single instrument run and bringing per-patient costs down. The key comes in bioinformatics, where the resulting per-patient data is examined against known sequences and pathogenic sequence variants for each of the genes examined. By doing this, oncologists can obtain a highly detailed picture of each of the points of dysregulation in a specific tumor; with luck, one or more of these may be the target (or immediately upstream of the target) for an available specific drug. Rather than treating all cancers as the same, oncologists now begin to have the ability to select a treatment which is customized to the unique root cause.
Already, the literature is beginning to include reports where use of an oncogene panel has identified that a cancer presenting as one type (based on cell type and appearance) is discovered at a genetic level to share commonalities with another very different cancer type—for which a specific drug is available. Informed by the oncogene panel results, use of a chemotherapeutic agent which would not otherwise have been considered has yielded highly effective treatments in these cases. While this opens complexities related to appropriate drug labeling—whether it should be more related to specific genetic defect, than to cancer class—the increasing use of oncogene panels and the concomitant development of yet more therapeutic agents targeted to specific pathways promise to provide significant advances in the treatment of cancers as highly individual presentations.

About the Author

John Brunstein, PhD
is a member of the MLO Editorial Advisory Board. He serves as President and Chief Science Officer for British Columbia-based PathoID, Inc., which provides consulting for development and validation of molecular assays.