The molecular menu: evaluating approaches to tumor profiling
To earn CEUs, visit www.mlo-online.com
LEARNING OBJECTIVES
Upon completion of this article, the reader will be able to:
LEARNING OBJECTIVES
Upon completion of these articles, the reader will be able to:
1. Define tumor profiling
2. Identify the importance of molecular profiling
3. Describe current methods for molecular testing
4. Define laboratory-developed tests (LDTs)
5. Identify the most prevalent uses for molecular testing.
Cancer is the second-leading cause of death in the United States, approaching parity to cardiovascular disease; there were more than 580,000 cancer deaths in 2013.1 There are approximately 230 known cancer types, and together they account for nearly one of every 4 deaths.2 While mortality rates by cancer type have fluctuated over the decades, the overall incidence and mortality rate is expected to remain relatively constant in the near future, with approximately 1.7 million new cancer cases and 590,000 deaths expected during 2015.2
Because of the diversity of cancer types and their concomitant spectra of characteristics, cancer remains a challenging disease to investigate and clinically manage. Despite this, the five-year survival rate across all cancers has doubled since 1950, and between 2003 and 2012, the overall cancer mortality rate has decreased by 1.5%.3 This is a result of the combination of earlier detection, greater understanding of factors that promote tumor growth, and improved treatment regimens over the years.
Historically, the treatment of cancer was performed by surgical removal of the tumor, but this was only effective if the cancer was small and localized enough to be completely removed. Later, radiation was used after surgery to control small tumor growths that remained. Then, chemotherapy was added to destroy small tumor growths that had spread beyond the reach of the surgeon and radiotherapist. This evolution in treatment occurred over the twentieth century and remains the standard of care for most cancers.
Recently, the development of targeted therapy for cancer treatment has provided physicians with the ability to design treatment around specific mutations that are unique to the tumor cells. This approach has demonstrated great promise but often requires a molecular profile of the tumor in order to determine whether the treatment may be effective.
This review will highlight the different approaches to molecular profiling of tumor cells, their advantages and drawbacks, and how they may fit into the current regimen for the treatment and/or diagnosis of cancer.
The importance of molecular profiling

Another example is the compound panitumumab (Vectibix), which was initially approved by the U.S. Food and Drug Administration (FDA) in 2006 for the treatment of EGFR-expressing metastatic colorectal cancer.6 A few years after its approval, subsequent studies showed that the effectiveness of panitumumab was greatly reduced in patients harboring KRAS mutations.7-9 This prompted the FDA in 2009 to update the indication and usage of panitumumab to recommend administration only when KRAS mutations were absent. Today, companion diagnostic assays for determining KRAS status are routinely applied to determine whether EGFR-inhibitors should be used in the treatment of advanced colorectal cancer.
To date, only one of 15 oncology-related treatments have progressed from preclinical studies to FDA approval, due to lack of clinical effectiveness or morbidity associated with an effective dose.10 However, there are now more than 500 targeted therapies in various stages of development, spanning more than 100 different biomarkers.11 Obtaining a molecular profile for a tumor is likely to become part of standard clinical practice for the treatment of cancer, as this will determine which targeted therapy should be used or avoided.
Genetic variation types
The most frequently cited genetic variations are single nucleotide variants (SNVs), which are also known as point mutations. SNVs result from a substitution at a single nucleotide due to errors during replication. They can occur spontaneously due to the chemical instability of purine and pyrimidine bases, or they can be induced by exposure to reactive chemicals and ionizing radiation.12 Depending on where the substitution occurs, SNVs may result in a change in the amino acid sequence of the encoded protein (missense mutation), a premature truncation of the protein (nonsense mutation), or, because of the degeneracy of amino acid codons, a non-coding change (synonymous, or non-coding mutation). Intronic SNVs, while not affecting the amino acid sequence, may also result in changes in expression and/or splicing of a particular transcript.13,14
Another class of variants is termed indels (insertion/deletion). Indels are typically characterized by an insertion or deletion involving one or a few nucleotides, but they can also occur as small duplications of consecutive nucleotides, or more complex mutations involving simultaneous deletions and insertions of one or a few bases. Depending on the length of the indel, they can result in the addition or subtraction of amino acids in the protein without disturbing the downstream amino acid sequence and are classified as “in-frame” insertions or deletions. Alternatively, indels can cause a “frameshift,” resulting in the alteration of amino acid codons downstream of the indel, which usually leads to the introduction of a stop codon and premature truncation of the protein.
The third class of genetic variants, termed structural variants (SVs), are large structural anomalies of DNA that result from breakpoints between different chromosomes or within a single chromosome. Occurring by inversion and/or translocation, these breaks can result in fusion genes and associated fusion proteins. Citing the earlier example, Philadelphia-positive cells are leukocytes that harbor a reciprocal translocation between chromosomes 9 and 22, resulting in the BCR-ABL fusion protein.
The final genetic variation type is copy number variations (CNVs), which include large duplications or deletions encompassing entire exons and affecting the functional domains of the protein. Sometimes these can include amplifications or deletions of an entire gene or region of a chromosome.
Types of molecular tumor testing
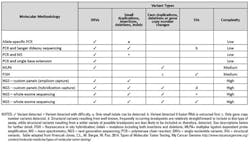
Different methods and analytes are used depending on the type of genetic variant under investigation, and each has its advantages and disadvantages in terms of cost, complexity, and discriminatory power. It is incumbent on clinical laboratory directors to educate themselves on the tools that are available for each desired application in the laboratory. Ideally, a lab leader selects a method that has the requisite analytical capability to identify the variant while minimizing the amount of effort and cost associated with the assay, but often a method will trade one advantage for another. Table 1 outlines some commonly used techniques to obtain genetic information from tumors, the type of variant detected, and the complexity of the method. Brief descriptions of the methodologies follow.
Allele-specific PCR
Allele-specific PCR is a variant form of real-time PCR in which short oligonucleotide probes, specific for the wild-type and variant sequence, are mixed together in the amplification step. Each probe is labeled with a different reporter, and following hybridization, the DNA polymerase extends the annealed probe, releasing it for detection. Subsequent PCR cycles result in amplified signals, allowing for measurement of one or both alleles of interest. Various allele-specific PCR assays have been approved as a companion diagnostic tool for the treatment of FDA-approved therapies, including assays that detect BRAF V600E, and EGFR exon 19/20 insertions/deletions and KRAS mutations.
This method has the benefit of employing commonly used PCR platforms, is fairly straightforward and fast to perform, and is inexpensive when interrogating a limited number of variants across a large cohort of samples. Despite these advantages, allele-specific PCR cannot detect large indels, copy number variations, or structural variants, although there are newer assays that utilize amplification across known DNA breakpoints in order to detect the presence of fusion transcripts (e.g. BCR-ABL). The most significant drawback of allele-specific PCR is that it is target-specific and requires a priori knowledge of a mutation. It cannot be used to detect mutations outside the known target of interest.
Mass spectrometry (MS)
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) can detect more than 100 variants simultaneously. These tests are custom-designed to interrogate tissues for mutations of interest and are usually designed to look for common mutations that occur at regions harboring frequent genetic variations. First, primer extension is performed using site-specific primers and a combination of dNTPs and ddNTPs, selected so that different alleles will result in products of different and predicted sizes. Primer extension products are then resolved by mass spectrometry in order to determine the genotype.
The two main advantages of MS are its sensitivity and scalability; it reliably detects minor allele frequencies approaching five percent to 10 percent while enabling the simultaneous interrogation of multiple SNVs. Like allele-specific PCR, MS is target-specific and cannot detect other mutations in tumor DNA that may be present. A further drawback is the dependence on mass spectrometry instrumentation, which is less common in biological laboratories and carries a higher capital cost.
Single-base extension assay
Single-base extension assays (SBEs) are performed by annealing a primer immediately adjacent to the variant of interest. After hybridization, the primer is extended by the polymerase with dideoxynucleotide terminators in the absence of dNTPs. Each of the four terminators is labeled with a unique reporter so that it is possible to identify the incorporated base. Typically, multiplex PCR reactions containing primers of differing length are used so that multiple targets can be resolved using capillary electrophoresis or mass spectrometry.
Similar to MS, single-base extension has higher sensitivity while being able to resolve multiple variants at once. This assay also does not require any specialized equipment. However, when using a non-commercial kit, custom assays must be designed and tested for each target and cannot detect mutations outside the target design.
Multiplex ligation-dependent probe amplification (MLPA)
MLPA is a preferred method to screen for deletions or duplications of multiple exons across a gene. For each target, custom primers (probes) are annealed adjacently so that they are in a position to be ligated. Each probe contains a unique tail that does not hybridize to the target. These tails are used as a template for the subsequent amplification of the target region. Due to the unique tail lengths, PCR fragments of different sizes are generated and resolved/quantitated using capillary electrophoresis or other methods.
Commercial assays are available for MLPA and do not require any specialized equipment. However, if commercial assays are not available for targets of interest, then customization and testing is required. This method is fast and relatively inexpensive, but like the assays mentioned before, it cannot identify variations outside the target design.
Fluorescence in situ hybridization (FISH)
FISH is a widely used hybridization technique used to assess copy number changes and structural variations. Fluorescent DNA or RNA probes are constructed such that they hybridize to chromosomal regions of high sequence complementarity. Fluorescence microscopy is then used to determine where the fluorescent probe is bound to the chromosomes. By designing probes specific to known chromosomal breakpoints and quantifying the fluorescent signals as compared to a standard, fusion gene products, gene expression, and copy number variation can be detected.
While this method readily detects gene copy number changes and targeted SVs that are not as easily detected by other methods, it lacks nucleotide resolution and cannot detect most types of mutations occurring in solid tumors. It can also be a relatively expensive procedure to perform.
DNA or RNA sequencing
The last method in this review is DNA or RNA sequencing. Most traditional sequencing, or Sanger Dideoxy sequencing, is performed on PCR products. Sequencing primers are annealed to the PCR product and extended by DNA polymerase, dNTPs, and a mixture of fluorescently labeled ddNTPs. Each of the four ddNTPs is labeled with a different fluorescent dye. Random incorporation of the labeled ddNTPs results in termination of strands at each location along the sequence. The reaction primers can be extended by the polymerase by approximately 1,000 nucleotides before the signal-to-noise ratio threshold is exceeded. Capillary electrophoresis separates the strands by size, and the terminating nucleotides are measured using fluorescence spectroscopy. In a clinical laboratory, both the forward and reverse strands are typically sequenced.
Unlike the approaches previously mentioned, Sanger sequencing can be used to detect unknown mutations and has a high accuracy rate. It can detect a variety of mutations, such as indels, SNVs, and even gene fusions if RNA from the fusion transcript is obtained. However, this method can be labor-intensive, and the sensitivity for variation detection has been established at 20% minor allele frequency. Sanger sequencing was the workhorse of the human genome project and is now being widely used in clinical laboratories to confirm variants identified by next generation sequencing.
A newer method of sequencing is gaining wide acceptance in clinical laboratories. NGS, or next-generation sequencing, is a method by which DNA or RNA fragments are sequenced in a massively parallel fashion. After nucleic acid substrate is obtained, short adaptor sequences are added to the fragments. These adaptors contain a universal priming sequence motif so that a single primer can be used to extend the template of every fragment in parallel during the sequencing reaction. Each fragment, now called a library molecule, is separated and clonally amplified on a solid surface such that its physical location is recorded. By a variety of mechanisms, sequential sequencing reactions are performed, and the collective signal from the clonally-amplified library molecules is used to build a sequence read. Since the coordinates of each amplified molecule have been determined, the sequence read is assembled as the sequencing reactions progress. After completion, the sequence reads (which typically number in the millions) are aligned to a reference genome to determine which variants might be present.
NGS can be performed on multiplex PCR products, whole genomes, or regions of the genome that have been isolated and enriched by various methods. This type of sequencing has been shown to reliably detect SNVs, indels, and copy number variants, and more recent work in RNA sequencing has established the ability to detect fusion transcripts.15 While an extremely powerful tool, NGS is expensive and complex compared to PCR-based methods and requires robust IT capabilities in order to analyze and manage the large amount of data that is produced.
The last two decades have seen rapid growth in the molecular tools available to perform tumor profiling. While the development of these molecular tools and the rapid pace of targeted therapy are reasons to embrace the concept of personalized medicine, practitioners must temper this enthusiasm with the proper understanding of the strengths and weaknesses of each molecular technique. Whenever possible, the most cost-effective choice of technologies and methods should be used so that economic efficiency and analytical validity are maximized. As molecular profiling methods become more sophisticated, the need for interpretative tools and reporting of results in a clear and understandable way to practicing oncologists will be paramount. There is little doubt that precision-guided medicine is on the horizon. It’s the responsibility of the medical community to harness the appropriate tools to achieve the best possible outcomes for patients and society.
References
- Kochanek KD, Murphy SL, Xu J, Arias E. Mortality in the United States, 2013. NCHS Data Brief. 2014;178:1-8.
- American Cancer Society: Cancer Facts and Figures 2015. Atlanta, GA: American Cancer Society, 2015. http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf. Accessed May 13, 2015.
- Howlader N, Noone AM, Krapcho M, et al (eds). SEER Cancer Statistics Review, 1975-2012, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2012/. Based on November 2014 SEER data submission, posted to the SEER web site, April 2015. Accessed May 13, 2015.
- Deininger MW, Druker BJ. Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacol. Rev. 2003;55(3): 401-423.
- Leukemia—Chronic Myeloid—CML: Statistics | Cancer.Net. http://www.cancer.net/cancer-types/leukemia-chronic-myeloid-cml/statistics. Accessed May 19, 2015.
- U.S. Food and Drug Administration. Panitumumab license letter of approval. http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2006/125147LTR.pdf. Accessed May 19, 2015.
- Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26(3):374-379.
- Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626-1634.
- Douillard, J-Y, Oliner, KS, Siena, S. Panitumumab–FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. NEJM. 2013; 369 (11):1023–1034.
- Thomas, D. Oncology Clinical Trials – Secrets of Success. 2012. http://www.biotech-now.org/business-and-investments/2012/02/oncology-clinical-trials-secrets-of-success#.Accessed May 19, 2015.
- Oncology Therapeutics Market to 2017. GBI Research, December 2011.
- Lodish H, Berk A, Zipursky SL, et al. Molecular Cell Biology. 4th edition. New York: W. H. Freeman; 2000; Section 8.1. Mutations: Types and Causes.
- Cech TR. Self-splicing of group I introns. Annu Rev Biochem. 1990;59:543–568.
- Michel F, Ferat JL. Structure and activities of group II introns. Annu Rev Biochem. 1995;64: 435–461.
- Yoshihara K, Wang Q, Torres-Garcia W, et al.. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2014 doi:10.1038/onc.2014.406 (Epub ahead of print).
With MDx here, what are the implications for LIS?
By Megan Schmidt, BSc
Clinical laboratories are a profitable resource for hospitals, and they can also be a strong asset as hospitals look to grow their physician relations and deepen their footprint in the local community. As hospital systems compete, however, clinical labs are increasingly being asked to do more with less. Many hospital labs are turning to molecular testing to grow and optimize outreach investment, increase penetration in the community, and add profitability. Currently, molecular testing comes with reimbursement challenges. That may change in the years ahead, but even today, molecular diagnostics (MDx) can offer higher payments and provide a competitive edge not only over other community hospitals but also independent labs.
Molecular diagnostics is also, by far, the fastest growing segment of lab testing. This is due to a boom in molecular disease markers and assays, a decrease in assay and instrumentation cost and complexity, and the use of laboratory-developed tests (LDTs). Molecular testing can be used for diagnosis, prediction, treatment, prevention, and drug research/clinical trials. It can include infectious disease testing, as well as oncology, forensic, paternity, and phamacogenomics testing. An increasing number of hospitals are looking to bring these tests in house to reduce cost, improve turnaround time, and build their lab’s value.
Molecular test samples can start as clinical pathology samples (blood or urine) or anatomic pathology samples (tissue or cyto fluids). There are several procedural steps that a sample must typically move through before being resulted. Each of these procedures can be discrete processes with instrument interfaces, QC, and reagents associated. Often the result is an interpretation of or comment on the findings that is achieved through integration of information.
Some in vitro diagnostics (IVD) tests have been approved by the U.S. Food and Drug Administration (FDA). Many labs modify these tests or create LDTs and validate these assays internally. Eighty-five percent of labs run at least one LDT, and most genetic tests are done as LDTs.1 Those tests are covered by Clinical Laboratory Improvement Amendments (CLIA), which define most basic quality systems for clinical laboratories. However, these regulations provide no specific guidance on molecular diagnostic testing. Molecular laboratory directors are faced with the task of establishing quality assurance programs that are consistent with the mission and scope of their respective laboratories.
The Clinical and Laboratory Standards Institute (CLSI) published MM3-A2-Molecular Diagnostic Methods for Infectious Diseases as approved guidelines to be used by clinical laboratories for the field of molecular microbiology, which uses nucleic acid methods to diagnose and manage patients with infectious diseases. An additional publication, CUMITECH 31 Verification and Validation of Procedures in the Clinical Microbiology Laboratory, provides guidance on producing consistent, accurate and precise results in the clinical microbiology laboratory. College of American Pathologists (CAP)-accredited laboratories can utilize the CAP Molecular Pathology Checklist and the molecular microbiology section of the Microbiology Checklist to assess compliance with CAP rules and regulations to prepare for CAP inspections. The CAP checklists reference both CLIA ’88 and CLSI publications.
LDTs add special workflow considerations because the QC, usually managed by FDA-approved kit providers, must be maintained in the lab, and this requires much more record- keeping around standards and reagents. Adding to concern about QC, the FDA intends to increase guidance on LDTs. Some clients manage this workflow outside the LIS and store molecular results on paper or homegrown applications, but many are looking for more formalized workflow support to replace the paper-based manual processes currently used.
Molecular testing requires a flexible, protocol/procedure-based laboratory workflow engine with strong QC and inventory controls. The clinical LIS must move a molecular test from one procedure to the next, with interfaces and QC as required per procedure, to the final result based on a protocol. Clinical LIS providers are stepping up to fill gaps around inventory and QC controls, with the advantage of an integrated clinical system. Clinical laboratories are investing heavily in molecular platforms that can detect and quantify infectious disease agents, evaluate the genetic basis of disease, and inform treatment. When evaluating an LIS, lab decision makers should consider the system’s ability to support molecular-based testing and genetics.
Infectious disease is one of the most prevalent molecular testing segments. Within Infectious Disease labs, molecular testing is used to diagnose with pathogen identification and to support treatment with antibiotic susceptibility and toxin information. Often, it is also used to support public health; the isolate can be used to help identify the source during an outbreak. In individual cases and in pandemics, time is always of the essence, but precision and quality cannot be sacrificed.
Consider the impact of:
- Molecular testing on an individual case: Mass spectrometry (MS) can provide the ability to report an organism to a clinician in minutes. The shorter time reduces assumptions and allows for more effective treatment by getting the right drug the first time.
- Improvements on the overall cost of treatment: At Laboratory Confab in 2014, Henry Ford Hospital presented that MALDI-TOF MS-based Blood Culture reaped an annual lab savings of over $100,000.2 The savings to the health system could also be calculated in reduced length of stays related to faster negative results.
- Preventing an epidemic: At the 2013 G2 Molecular conference, University of Arizona Medical Center presented a case study in which the seven-day turnaround time for a pertussis culture resulted in 94 exposures requiring prophylaxis, at a cost of $44,000.3 A molecular PCR-based assay can return results the same day and reduce the risk of an outbreak.
- Public health in general: Public Health England is applying whole genome sequencing for pathogen identification.4 NGS is one of the most critical MDx technologies from a market perspective and is being applied to microbiology.
- The field of clinical microbiology is undergoing rapid change, with novel molecular technologies cutting the time needed to diagnose infections, enabling more precise and appropriate individualized therapies, improving patient care, and optimizing clinical outcome. Clinical laboratory leaders should evaluate the molecular and LDT solutions offered through their LIS provider. They may be surprised to find how this growing and important field of diagnostics can be streamlined.
References
- Department of Health and Human Services. Secretary’s Advisory Committee on Genetics, Health, and Society. U.S. System of Oversight of Genetic Testing: A Response to the Charge of the Secretary of HHS. http://osp.od.nih.gov/sites/default/files/SACGHS_oversight_report.pdf. Accessed May 19, 2015.
- Zarbo RJ, Sharma G. Coordinating Clinical Laboratory and Anatomic Pathology Services in Today’s Integrated Clinical Care Continuum. Lab Quality Confab 2014. http://www.labqualityconfab.com/wp-content/uploads/ZARBO-SHARMA.tue_.9.30am.FINAL-V3.pdf. Accessed May 19, 2015.
- Whitfield NN. An ever-changing environment: molecular. G2 MDX 2013. Clinical Genomics Software Short Course. Bio-IT World April 2013.
- Underwood A. Developing and provisioning robust automated analytical pipelines for whole genome-based public health microbiological typing, BioIT Conference. April 2015.
