
As next-generation sequencing (NGS) is expanding into a wider range of applications, laboratories face constant challenges that they need to tackle to fully benefit from the power of NGS. In this article we discuss some of these challenges—as well as innovative ways of addressing them to enable reliable analysis of a wider range of variants and samples.

Archived tissue biopsies
When studying the development and progression of cancers, archived tissue biopsies are a valuable resource. However, these samples are typically stored as formalin-fixed, paraffin-embedded (FFPE) blocks—a process that can severely damage DNA. When older samples are used for analysis, it is likely that the integrity of the DNA has deteriorated even further. Low-quality DNA limits a sample’s suitability for analysis by NGS, and can make distinguishing true, low-frequency mutations more difficult.
To improve the likelihood of success when analyzing FFPE-derived DNA, one can utilize a hybridization-based target enrichment approach. This enrichment method relies on random shearing of DNA followed by selective capture of relevant fragments. It provides superior tolerance to the effects of fragmented DNA compared to amplicon-based approaches.
Further improvements in sequencing quality, and subsequent results, with FFPE samples can be made by including a DNA pre-treatment in the library preparation workflow. DNA pre-treatment repairs certain types of DNA damage, such as cytosine deamination, nicks and gaps, oxidized bases, and blocked 3’ ends.
To investigate the effect of DNA repair with formalin-compromised samples, the performance with increasing levels of DNA damage was compared—both with and without repair. DNA repair resulted in significant improvements in DNA integrity, resulting in a considerable increase in pre-capture library yields and a subsequent improvement in mean target coverage (Figure 1). The results show that hybridization-based enrichment can enable high sequencing coverage—even with low input amounts of severely damaged DNA.
Overcoming technical limitations
In the detection of rare genomic variants, molecular techniques such as digital droplet PCR (ddPCR) and Sanger sequencing, though well established, have limitations. ddPCR is hampered by covering only one known mutation per assay and offering limited possibilities for multiplexing, while Sanger sequencing offers limited sensitivity, restricted to 20 percent allele frequency (AF). These issues can be overcome by the use of targeted sequencing panels offering multi-gene analysis at high sensitivity. The following case study from a large UK hospital demonstrates the benefits that targeted NGS can bring to the laboratory.
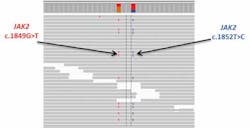
When analyzing myeloproliferative neoplasm (MPN) samples, ddPCR was routinely used to identify and quantify the common V617F mutation in JAK2. If the test was negative, follow-up analysis with Sanger sequencing and capillary electrophoresis was needed to look for alternative causative mutations.
This particular case was complicated by the presence of a rare second mutation only three bases upstream (c.1852C>T). As a result, the V617F mutation was not detected because of this second variant under the primer binding site, resulting in a false negative result. Using a custom hybridization-based myeloid NGS panel, both mutations were detected at an AF of 32 percent (Figure 2). Although rare, this case highlights the accuracy and precision of a targeted sequencing approach in identifying mutations that could not be picked up otherwise.
Hard-to-sequence regions
As comprehensive analysis of genes or exons is a key strength of NGS, it is important that NGS methods provide good uniformity of coverage to ensure that variants are detected with as high a degree of certainty as possible. A common challenge that can negatively affect coverage uniformity is the presence of hard-to-sequence genomic regions, such as internal tandem duplications (ITDs) and regions with high GC or AT content. These phenomena occur in many genes, for example CEBPA and FLT3 in acute myeloid leukemia (AML).
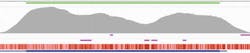
FLT3 contains several ITDs that can be long and are often masked, making it hard to obtain consistent, high coverage across the gene. However, when using hybridization-based target enrichment, intuitive modifications to the bait design makes it possible to bait right up to the repeat region. This enables high-coverage sequencing of the repeat region and therefore clear detection of the ITD itself (Figure 3a).

Several key mutations that can lead to AML are linked to the CEBPA gene. NGS analysis of this gene is challenging due to areas with high GC content across the gene as well as the presence of several repeat regions. When coverage of these areas is too low, they may require supplementary fill-in with Sanger sequencing. Again, an optimized bait design in terms of sequence and bait length—in conjunction with a hybridization-based targeted sequencing approach—ensures high-quality, uniform coverage in these regions, minimizing the requirement for supplementary analysis (Figure 3b).
Copy number variation
Combining the detection of point mutations with copy number variation (CNV) can result in significant time savings in laboratories. However, detecting CNVs reliably from NGS data is only possible with deep coverage that is highly uniform across all target sites. Achieving this can often be expensive and time-consuming. Furthermore, due to the size and complexity of the dataset, there are also software challenges that need to be considered to ensure effective CNV calling.
A good example of the benefits that can be achieved by combining the detection of point mutations and CNV is the analysis of the LDLR gene, which is involved in familial
hypercholesterolemia (FH). FH samples are often characterized by multiple point mutations in LDLR as well as intragenic CNVs in 10 percent of cases.
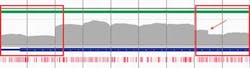
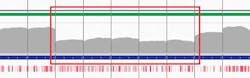
The analyses in Figure 4 show sequencing results of two LDLR genes with confirmed CNVs. They clearly show a single heterozygous deletion in one sample (Figure 4a) and a heterozygous deletion across two regions with a mid-exon breakpoint in the other (Figure 4b). This demonstrates how targeted sequencing can reliably detect CNVs, enabling combined point mutation and CNV detection in a single assay.
Overcoming challenges
NGS has transformed the sequencing landscape with ever-increasing speed, precision, and affordability. Overcoming challenges is the key to expanding NGS into new applications. When using targeted sequencing to study specific regions of the genome, the deep, uniform coverage of hybridization-based enrichment enables reliable analysis in a range of demanding sequencing applications, such as FFPE-derived DNA, discovering rare mutations, detecting CNVs, and studying challenging areas of the genome.
