Hemoglobinopathies and clinical laboratory testing



To earn CEUs, see current test at
www.mlo-online.com
under the CE Tests tab.
LEARNING OBJECTIVES
Upon completion of this article, the
reader will be able to:
- Describe characteristics of hemoglobinopathies.
- Identify measurements important for classification of hemoglobin
variations. - Describe methods available for defining hemoglobin variants.
- Describe the role of neonatal screening for detection of hemoglobin
defects.

Hemoglobin (Hb) is
the oxygen-carrying protein of erythrocytes. It is an iron-containing,
tetrameric metalloprotein that consists of two pairs of unlike globin
chains (i.e., two “-type and two “-type globin chains). The globin
chains form a shell around a central cavity containing four heme
prosthetic groups, each covalently linked to a single chain. The heme
found in Hb is a porphyrin ring bound to a central iron atom, which
serves as the site of oxygen binding. The “-type globins are encoded by
a gene cluster on chromosome 16 that contains the embryonic ” gene and
two adult ” genes in series, oriented in the 5-prime (5′) to the 3-prime
(3′) direction (see Figure 1A). The “-type globin genes are clustered on
chromosome 11 (see Figure 1A), and include the embryonic “, two tandem
fetal ” genes, and the adult ” and ” genes oriented in the 5′ to 3′
direction. The globin genes are activated from 5′ to 3′ during embryonic
and fetal development. In the healthy neonate, fetal Hb, or Hb F (“2“2),
is the major species (~70%) (see Figure 1B). Hb F is replaced by the
major and minor adult Hbs, Hb A (“2“2) and Hb A2
(“2“2), during the first 6 to 12 months of life
(see Figure 2). Thus, in healthy adults, Hb is composed of Hb A (~95%)
and Hb A2 (~3.5%), with only trace amounts of Hb F. The normal diploid
cell produces Hb A from four ” and two “-globin genes. The “- and “-globin
chains consist of 141 and 146 amino-acid residues, respectively.
Hemoglobinopathies are inherited single-gene
disorders that affect Hb production and function. It is estimated that
around 7% of the world population carries a globin-gene mutation; and in the
majority of cases, it is inherited as an autosomal recessive trait.1
Hemoglobinopathies can be classified broadly as qualitative and quantitative
disorders. Qualitative hemoglobinopathies result from “- or “-globin gene
mutations that cause structural alterations in the Hb molecule. Quantitative
hemoglobinopathies, or thalassemias, arise from mutations that cause
decreased synthesis of otherwise normal “- or “-globin chains, resulting in
stoichiometric imbalance of the subunits. More than 700 structural Hb
variants and thalassemias have been described in published scientific
journal articles2; and, as of June 19, 2009, 1,361 entries have
been posted on HbVar, a database on human Hb variants and thalassemias
available online at
http://globin.cse.psu.edu/hbvar/menu.html .
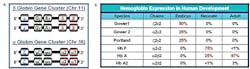
Figure 1. Hemoglobin. A) Gene clusters for
hemoglobin on chromosome 11 and 16.
B) Hemoglobin expression in human development.
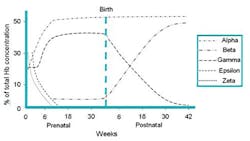
Figure 2. Prenatal and postnatal time dependant expression of different hemoglobins forms as a percentage of Hb concentrations.
Most hemoglobinopathies are clinically benign; and in
most cases, not enough scientific evidence has been demonstrated to prove or
disprove any pathological effect. The convention used by HbVar to
characterize structural variants includes the affected globin gene, altered
residue number, protein domain, and amino-acid substitution. Thus, for
example, Hb S is characterized as ” 6(A3) Glu>Val to indicate that a”-globin
mutation at residue 6, in domain A3, causes an amino-acid change from
glutamic acid to valine. This change results in Hb tetramers that aggregate
into arrays upon deoxygenation in the tissues, leading to the deformation of
red blood cells (RBCs) into sickle-like shapes, making them relatively
inflexible and unable to traverse the capillary beds. Among the structural
variants, a high gene frequency and significant clinical or hematological
effects are observed with Hb S, Hb C (” 6(A3) Glu>Lys) and Hb E (“26(B8) Glu>Lys).
In the case of Hb C, the glutamic-acid-to-lysine substitution leads to an
unstable Hb that precipitates in red blood cells to form crystals, thus
decreasing the deformability of red blood cells. Affected erythrocytes are
removed by the spleen. In the case of Hb E, the mutation at codon 26 of the
“-globin gene causes a substitution of glutamic acid by lysine and also
activates a cryptic mRNA splice site, resulting in the reduced synthesis of
the “-E chain, leading to a thalassemia phenotype.3
Hb S is the variant that causes sickle-cell disease
(SCD) (OMIM database No. 603903). SCD includes a group of genetic disorders
characterized by chronic hemolysis and episodic vascular occlusion.1
The disorders are found primarily in people of African, Mediterranean, and
Southeast Asian ancestry.4 Vascular occlusion results in episodes
of severe pain and tissue infarction, while the consequences of hemolysis
include chronic anemia, jaundice, predisposition to aplastic crisis,
cholelithiasis, and delayed growth and sexual maturation. The condition can
result in acute and chronic injury to most of the organs, in particular, the
spleen, brain, lungs, and kidneys.5 Sickle-cell anemia (Hb SS)
accounts for 60% to 70% of sickle-cell disease in the United States.6
Other forms result from coinheritance of Hb S with other abnormal “-globin-chain
variants. The most common of these forms includes sickle-Hb C disease (Hb
SC),1 a condition that occurs in about one in 835
African-American births and in about one in every 25,000 births in the
general population, as well as two types of sickle “-thalassemia, Hb S
“/zero-thalassemia and Hb S “/plus-thalassemia, which together occurs in
about one in every 50,000 births. Those with Hb S “/zero-thalassemia usually
have a severe form of the disease. People with Hb S”/plus-thalassemia tend
to have a milder form of the disease. Sickle-cell trait refers to situations
where the individual is a sickle-cell carrier and is asymptomatic.
Because of this significant reduction of morbidity and
mortality, neonatal screening for sickle-cell disease and other
hemoglobinopathies has become standard practice in the United
States.
Reduction in the amount of the normal globin chain
produced is characteristic of thalassemias. The clinical manifestations
range from mild anemia with microcytosis (thalassemia trait) to fatal severe
anemia (Hb Bart’s hydrops fetalis7 or “-thalassemia major). The
two main types of thalassemia are named according to which adult globin gene
is dysfunctional: “- and “-thalassemia.8
The decreased globin-chain synthesis may result from gene deletion or from
mutations that adversely affect the transcription or stability of mRNA
products. Disease is caused by insufficient functional hemoglobin, as well
as tetramer formation of the unaffected globin chain. In “-thalassemia,
tetramers of adult ” chain (named Hb H) and fetal ” chain (named Hb Bart’s)
are unstable in erythrocytes and cause hemolytic anemia. In “-thalassemia,
tetramers of ” globin are unstable in erythrocytes and bone-marrow
erythroblasts, resulting in hemolytic anemia and intramedullary cell death.
In severe cases, massive erythroid hyperplasia in the marrow causes bone
deformities. The great majority of “-thalassemia cases are caused by large
deletions of one or both “-globin genes (HBA1 and HBA2) on chromosome
16 (see Figure 1A). Single-gene deletions of HBA1 or HBA2 are prevalent in
many areas of the developing world. In contrast, large deletions
encompassing both HBA1 and HBA2 are most common in Southeast Asia, and are
very rare in patients of African ancestry. The “-globin deletions can be
inherited as homozygous or heterozygous defects, resulting in loss of one to
four ” globin genes. Severity of disease is dependent on the extent of gene
deletion:
- Loss of one “-globin gene is clinically and hematologically silent.
- Loss of two genes results in “-thalassemia trait, characterized by
microcytosis with little or no anemia. Two-gene deletion can occur when
both genes on a single chromosome 16 are lost in the patient’s genome
(“deletion in cis”), or when a single gene is lost on each version of
choromosome 16 in the patient’s genome (“deletion in trans”). - Loss of three genes results in Hb H disease, a moderate hemolytic
anemia. - Loss of all four genes is incompatible with independent life.
- Death occurs in utero from Hb Bart’s hydrops fetalis.
In contrast to “-thalassemia, “-thalassemia is
characterized by small missense or nonsense mutations in the “-globin gene
(HBB), which reduce or completely abrogate gene expression. Mutations that
eliminate expression are labeled “-zero, while those that reduce expression
are labeled “-plus. Severity of this condition is dependent on the extent of
“-globin chain underexpression. Heterozygous inheritance of a “-thalassemia
mutation results in a trait condition, sometimes termed “-thalassemia minor.
Like “-thalassemia trait, this condition is characterized by microcytosis
with little or no anemia, although it is distinguished by increased Hb A2
production. Homozygous inheritance results in “-thalassemia major (absent Hb
A production) or “-thalassemia intermedia (severely reduced Hb A
production), which are associated with moderate to severe hemolytic anemia
Diagnostic recommendations regarding the laboratory
investigation of these conditions were first made in 1975 by the
International Committee for Standardization in Hematology expert panel on
abnormal Hbs and thalassemias. The recommended initial testing included a
complete blood count, or CBC, electrophoresis at pH 9.2, tests for
solubility and sickling, and quantification of Hb A2 and Hb F.2
The identification of an abnormal Hb required further testing, using
addition techniques such as electrophoresis at pH 6.0 to 6.2, globin-chain
separation, and isoelectric focusing (IEF). Heat and isopropanol stability
tests were recommended for detection of unstable Hbs or Hbs with altered
oxygen affinity.2 Although electrophoresis at alkaline and acid
pH has been widely used for many years, the emergence of cation-exchange
high-performance liquid chromatography, or HPLC, as the method of choice for
quantification of Hb A2 and Hb F and identification of Hb variants,9
streamlined the recommended preliminary and follow-up tests for the
identification of hemoglobinopathies and thalassemias, and provided rapid
and complete diagnostic work-up in a majority of cases. The elements of
choice include a CBC, Hb H test, ferritin, HPLC for Hb A2 and F
quantification, and detection of any Hb variants followed by electrophoresis
at both alkaline and acid pH.
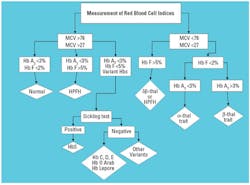
Measurement of hematological indices
The
hematological profile consists of measurements of the RBC indices and
includes Hb concentration, hematocrit, RBC number, mean corpuscular Hb
(MCH), mean cell volume (MCV), and red-cell distribution width (RDW, an
indicator of RBC size variation). Routinely, a blood film accompanies
the RBC indices. Structural hemoglobinopathies may have an impact on the
red-cell indices, and red-cell indices are critical to the differential
diagnosis of thalassemias. The general classification of thalassemias is
hypochromic and microcytic anemias; therefore, the MCV can be considered
as a key diagnostic indicator. Virtually all automated hematology
analyzers now provide a measurement of MCV that is both precise and
accurate.
Thalassemic individuals have a reduced MCV, and an
MCV of 72 femtoliter (fL or 10-12 liters) is maximally sensitive
and specific for presumptive diagnosis of thalassemia syndromes.10
The RDW can be used to differentiate between microcytic anemias, most
notably iron deficiency (increase in RDW), and the thalassemias, which — in
contrast — tend to produce a uniform microcytic red-cell population without
a concomitant increase in RDW.2 RDW may provide complementary
information but is not useful as a lone indicator. The RBC count is also
useful as a differential tool because the thalassemias produce a microcytic
anemia with an associated increase in the RBC number, while the other causes
of microcytic anemia, including iron deficiency and anemia of chronic
disease, are more typically associated with a decrease in the RBC number
that is proportional to the degree of decrease in Hb concentration. The Hb
concentration can provide complementary information since it is typically
decreased in thalassemia. The thalassemia-minor conditions produce minimal
decrements in the Hb concentration; whereas, thalassemia intermedia and
thalassemia major may be associated with moderate to severe decreases in Hb
concentration.2
Hb H inclusions

Figure 4. CAP survey results for Hb methods. Adapted CAP 2009 HB-A Proficiency Survey.
Hb H, an insoluble tetramer comprising four “-globin
chains, arises in the setting of “-thalassemia where the decreased
production of “-globin chains leads to “-globin excess. These Hb H
tetramers, when oxidized in vitro, precipitate and, hence, can be
visualized microscopically. Staining unfixed cells with an oxidative dye
such as new methylene blue or brilliant cresyl blue generated Hb H
inclusions. Because batch-to-batch variability in the dye occurs, positive
and negative control slides are critical.11 The Hb H stain is
non-specific in that other nucleic-acid or protein precipitates are also
stained. Hb H inclusions might be confused with reticulin and Howell-Jolly
bodies. In the case of Hb H disease, a disorder in which three of four “-globin
chain genes are non-expressed, 30% to 100% of red cells contain typical
inclusions; in contrast, “-thalassemia minor may be associated with as few
as one inclusion-containing cell in 1,000 to 10,000 cells. The absence of Hb
H inclusions, therefore, does not exclude thalassemia trait, but the
presence of typical inclusions may be helpful in confirming a presumptive
diagnosis.
The identification of Hb Bart’s in Asian infants can have
important genetic implications for couples [who] … may be at risk
for subsequent pregnancies complicated by hydrops fetalis.
Electrophoretic analytical methods
Electrophoresis has been the method of choice in
traditional hematological laboratories for qualitative and quantitative
analyses of the various Hb fractions. Cellulose-acetate electrophoresis
at alkaline pH (8.2 to 8.6), and citrate-agar or agarose-gel
electrophoresis at acid pH (6.0 to 6.2) provide a clear background,
allowing for the separation of the major Hbs (i.e., Hb A, Hb F, Hb S/D,
Hb C/E/O-Arab) and a number of less common Hb variants by densitometric
scanning.12
Visualization of the Hb bands is made possible by staining with amido
black and acid violet (or similar stains). The electrophoretic migration
of Hb C, Hb E, Hb A2, and Hb O is similar at alkaline pH. Hb S, Hb D,
and Hb G also co-migrate. The electrophoretic separation of Hb C from Hb
E, and Hb O and Hb S from Hb D and Hb G is accomplished at acidic pH. It
is not possible to differentiate between Hb E and Hb O, and Hb D and Hb
G using electrophoretic methods.2 Because of its simplicity,
cellulose-acetate electrophoresis remains one of the most popular
methods for Hb screening. In addition to being laborious, however,
electrophoretic techniques have the disadvantage of inaccurate
quantification of low-concentration Hb variants (e.g., Hb A2) or in the
detection of fast Hb variants (Hb H, Hb Bart’s). The precision and
accuracy of Hb A2 measurements using densitometric scanning of
electrophoretic gels is poor, especially with the emergence of various
HPLC techniques.13
Capillary electrophoresis is also used for Hb
analysis. This methodology, offered by Sebia, utilizes liquid-flow
electrophoresis for applications in the clinical-diagnostic setting to
separate, detect, and quantify normal Hbs and Hb variants. Red cell
hemolysates can be automatically prepared on the instrument. Samples are
then loaded onto the capillary, migration occurs, and relative
quantification and presumptive identification of the Hb fractions can take
place. According to the College of American Pathology (CAP) 2009 HG-A
proficiency surveys, 7% of clinical laboratories reporting
hemoglobinopathies use capillary electrophoresis.
Isoelectric focusing
Isoelectric focusing (IEF) on agarose gels is an
electrophoretic technique with excellent resolution and can be used to
separate different Hb fractions and variants and globin chains.14,15
IEF is frequently the first analytical test used for the diagnosis of Hb
fractions. If a better resolution is required, polyacrylamide gels can
be used instead. IEF allows the separation of Hb variants with
isoelectric points that differ by as little as 0.02 pH units. IEF allows
for more precise and accurate quantification than standard
electrophoresis due to the narrow bands obtained. IEF is an equilibrium
process in which Hb migrates in a pH gradient to a position of 0 net
charge. The Hb migration order of IEF is the same as that of alkaline
electrophoresis with resolution of Hb C from Hb E and Hb O, and Hb S
from Hb D and Hb G. In addition, Hb A and Hb F are clearly resolved.
Capillary isoelectric focusing
Capillary IEF (CIEF)16-18 is a
technique that combines the sensitivity of capillary electrophoresis
with the automated sampling and data acquisition of HPLC. Many published
works have described the utility of CIEF in the detection and
quantification of Hb variants; however, its use in the clinical
laboratory is limited. Separation of the Hb in this method is related to
the isoelectric point of the Hb. CIEF has been used to quantify Hb
variants Hb A2 and Hb F.16

Chromatographic analysis of Hbs and globin chains
Chromatographic methods are also widely used for
Hb quantitation and initial screening of Hb variants. Cation-exchange
HPLC has become the method of choice for the initial screening of Hb
variants19
(including neonatal screening where this is mandated) and for
quantification of Hb A2 and Hb F concentrations, and detection of
several abnormal Hbs. This method replaces electrophoretic techniques
for primary screening of Hbs of clinical significance, and is at least a
complementary tool for the presumptive identification of Hb variants.20
Bio-Rad Laboratories offers automated cation-exchange HPLC
instrumentation that has been widely used to quantify Hb A2, Hb F, Hb A,
Hb S, and Hb C. This HPLC technique suffers, however, from intrinsic
interpretive problems due to the fact that some Hb variants may co-elute
with Hb A2, hence, making quantification of Hb A2 impossible in those
cases.16,21 In individuals with Hb S, the presence of Hb S
adducts falsely increases Hb A2, thus complicating the quantification of
Hb A2 using cation-exchange HPLC. Recent advances in the cation-exchange
HPLC technology have resulted in the addition of columns and buffers
that alleviate this problem. In addition, using capillary-zone
electrophoretic method16 as well as micro anion-exchange
column methodologies, has been described22 that eliminates
this interference. According to 2009 CAP HB-A proficiency surveys,
ion-exchange HPLC, in which Hb species are separated based on charge
differences, accounted for the majority (93%) of the methods used for
the measurement of Hb and detection of hemoglobinopathies.
The quantification of Hb F is important in the
diagnosis of hereditary persistence of fetal Hb, juvenile chronic
myelogenous leukemia, and monosomy-7 syndrome, as well as for therapeutic
monitoring in patients with sickle-cell anemia. Immunodiffusion techniques
are laborious and relatively insensitive, and the densitometric scans of an
alkaline-electrophoretic gel cannot detect Hb F in healthy adults or in
those with marginally increased Hb F. Hb F quantities obtained by HPLC
techniques tend to be lower than from published Hb F quantities from
standard texts often are the result of alkali denaturation/spectrophotometry
methods.
In recent years, reversed-phase HPLC (RP-HPLC) of
globin chains has become an important tool for the study of Hb
abnormalities. HPLC has been used to diagnose thalassemia and
hemoglobinopathies, including detection of “-thalassemic genotypes in cord
blood.21 It has been used mostly to measure the “-globin chain
ratios in various Hb disorders, and, in addition, liquid chromatography/mass
spectrometry, or LC-MS, has been used to characterize other variant Hbs.23
Molecular diagnosis of hemoglobin-opathies
After presumptive identification of
hemoglobinopathies and thalassemia syndromes — and particularly for
purposes of genetic counseling — defining the mutation or deletion
present may be required. With the advent of polymerase chain reaction
(PCR), the array of diagnostic tools has been expanded. As with many
other genetic disorders, DNA amplification is coupled to a variety of
methodologies for detecting known mutations or screening for unknown
sequence alterations inside the human globin loci.
DNA is extracted from white blood cells for the
molecular diagnosis of thalassemias from chorionic villus samples and from
amniotic-fluid cells for prenatal diagnosis. Various low- ( samples/day), medium- ( throughput molecular-diagnostic techniques are available for genetic testing
of Hb disorders.12 Southern-blot hybridization of particular
restriction enzyme digests to labeled complementary gene probes is typically
used for the diagnosis of deletional mutations causing “-thalassemia
syndromes and some rare “-thalassemias. In addition, molecular methods for
detecting and typing the “-thalassemia deletions typically have required the
use of Southern-blot analysis. In general, laboratory diagnosis of “-/zero-thalassemia
carriers is performed by the HbH-inclusion body test (HbH prep).24
This test is laborious, observer-dependent, and reported to have poor
sensitivity. Multiplex PCR has been shown to be more effective for the
diagnosis of “-thalassemia than the HbH prep. It substantially increases the
sensitivity of the HbH prep for the detection of “-/zero-thalassemia. The
HbH prep, when used in conjunction with a low MCV, continues to have value
for the diagnosis of “-/zero-thalassemia in laboratories where PCR methods
are not available.24 Previous work has also demonstrated the
successful application of a gold nanoparticle-filled CE multiplex PCR method
for the diagnosis of “-thalassemia deletions. DNA containing “-thalassemia
deletions showed good agreement with results obtained by gel
electrophoresis.25 The identification of known globin-chain
mutations/deletions, including those for Hb S, E, D, and O, and several “-thalassemias
are achieved by PCR techniques using allele-specific probes after globin-gene
amplification, allele-specific primers, or deletion-dependent amplification
with flanking primers.26 When a deletion mutation is not
identified and there is high suspicion for a hemoglobinopathy, then sequence
analysis can be used to identify point mutations or sequence variation. In
addition, several PCR-based methods, including denaturing gradient-gel
electrophoresis and single-strand conformation polymorphism analysis, as
well as sequencing of the amplified globin gene DNA may be used for
identifying unknown mutations; however, DNA sequencing remains the ultimate
method for definitively identifying unknown sequence alterations.12
A recent study utilized multiplex ligation-dependent probe amplification
(MLPA) to analyze “-thalassemia patients from Southern China and concluded
that MLPA was a rapid and reliable method to determine the cause of both
deletional and non-deletional “-thalassemia.27
Diagnostic recommendations regarding the laboratory
investigation of these conditions were first made in 1975 by the
International Committee for Standardization in Hematology expert panel
on abnormal Hbs and thalassemias.
Neonatal screening for sickle-cell disease and other hemoglobinopathies
Neonatal screening for sickle-cell disease
receives the highest recommendation from the U.S. Preventive Services
Task Force (grade “A”), indicating high certainty of substantial net
benefit based on evidence from well-designed, well-conducted studies in
representative primary-care populations.28 Neonatal screening
is beneficial because presymptomatic diagnosis of sickle-cell disease,
followed by prophylactic penicillin therapy, has been shown to reduce
the incidence of pneumococcal sepsis by more than 80%.28,29
Ideally, prophylactic penicillin is started in infancy and is
implemented with pneumococcal and other vaccines, urgent management of
febrile illness, and family education about signs and symptoms of
splenic sequestration.30 Overall, the combination of newborn
screening and appropriate clinical follow-up has markedly decreased
childhood mortality from sickle-cell disease. For example, comparing
rates for 1999 to 2002 versus those for 1983 to 1986, mortality from
sickle-cell disease decreased by 68% at age 0 to 3 years (95% confidence
interval 58% to 75%) and by 39% at age 4 to 9 years (95% confidence
interval 16% to 56%).31
Because of this significant reduction of morbidity
and mortality, neonatal screening for sickle-cell disease and other
hemoglobinopathies has become standard practice in the United States. Some
statewide programs initially implemented targeted screening for high-risk
racial and ethnic groups, in particular infants of African, Mediterranean,
Middle Eastern, (East) Indian, Caribbean, and South and Central American
descent. These selective programs, however, experienced rates of missed
cases as high as 30%, as well as increased administrative costs and
litigation risk. Therefore, universal screening is recommended as most
efficacious and cost-effective by the American Academy of Family Physicians,
the American Academy of Pediatrics, and the American College of Medical
Genetics, and is currently implemented in all 50 United States, the District
of Columbia, and the U.S. Virgin Islands.28,29,32
Statewide screening programs analyze an eluate from
the dried blood spot that is obtained for tests of other congenital
disorders. The most common Hb testing methods are IEF and HPLC. Most
programs retest abnormal specimens with a complementary electrophoretic
technique, HPLC, immunologic tests, or DNA-based assays.33,34
Solubility tests are inappropriate for confirming sickle Hb in newborns,
because high levels of fetal Hb give false-negative results. The sensitivity
and specificity of current statewide screening systems are extremely high,
approaching 100%.28
Still, rare cases of hemoglobinopathy remain undetected at birth — even in
states with universal screening — mainly due to preanalytical errors that
include failure to offer testing, requisition or specimen mislabeling,
extreme prematurity with lack of adult Hb, and blood transfusion prior to
screening.29,33
By convention, Hbs identified by neonatal screening
are reported in order of expression level. At birth, normal infants
(homozygous AA) express a majority of fetal Hb (Hb F) and a minority of
adult Hb (Hb A) and, thus, would be characterized as Hb FA. By analogy,
infants with a homozygous hemoglobinopathy would be characterized as Hb
F-Variant (e.g., Hb FS for homozygous sickle-cell disease). Those with a
heterozygous hemoglobinopathy trait would generally be characterized as Hb
FA-Variant (e.g., Hb FAS for sickle-cell trait).
A range of sickle-cell disease variants can be
detected and differentiated by neonatal screening29,33 (see Table
1). Hb FS in infancy is compatible with several genotypes, implying a wide
range of future clinical severity. Most infants with FS-screening results
have homozygous SS, but other genotypes including sickle/”-zero thalassemia,
sickle/”-plus thalassemia, and sickle/delta-“thalassemia are possible. In
addition, compound heterozygosity for sickle Hb and hereditary persistence
of fetal Hb (S/HPFH) produces an FS phenotype at birth. Although relatively
uncommon, S/HPFH is very important to distinguish from sickle-cell disease,
since it is a benign trait condition. It can be suspected by an absence of
clinical and hematological effects, and by follow-up Hb analysis showing
persistent Hb F beyond 6 to 12 months of age. Newborn-screening algorithms
are capable of identifying sickle-cell-disease variations caused by compound
heterozygosity of Hb S and other mutant Hbs (e.g., FSC, FSC-Harlem,
FSD-Punjab, FSO-Arab), or by sickle/”-plus thalassemia with Hb S predominant
over Hb A (i.e., FSA).

All infants with Hb S detected by newborn screening should have confirmatory
tests of a second blood sample prior to 2 months of age, to allow early
detection of sickle-cell disease. Infants with Hb FS and other significant
sickle hemoglobinopathies should begin penicillin prophylaxis by 2 months of
age, and parents should be educated about the importance of urgent medical
evaluation and treatment for febrile illness, and for signs and symptoms of
splenic sequestration.29,33
Clinical and laboratory evidence of sickle-cell disease is rare before 2
months of age due to continued expression of fetal Hb, which inhibits
polymerization of sickle Hb. Postnatal suppression of Hb F occurs at a
variable rate in infants with sickle-cell disease. Therefore, some cases may
be difficult to distinguish from S/HPFH, in which case parental testing
and/or DNA analysis may be helpful if clinically indicated.29
Screening for other hemoglobinopathies and “-thalassemias
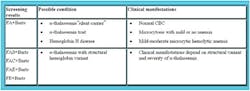
Neonatal screening can also identify infants with
non-sickle hemoglobinopathies, some of which may be severe and require
transfusion therapy29,33 (see Table 2). For example, infants
expressing only Hb F at birth may have the disabling condition “-thalassemia
major. This neonatal phenotype, however, may also be compatible with a
normal Hb genotype in premature infants with a lack of Hb A production.
Therefore, premature infants without Hb A need repeat testing to confirm
eventual production of adult Hb. Neonatal screening does not detect most
infants with milder “-thalassemia syndromes (i.e., “-thalassemia minor and
“-thalassemia intermedia), since some Hb A is produced. Infants with FE
require family studies, DNA analysis, or repeated hematologic evaluation
during the first one to two years of life to differentiate homozygous Hb E,
which is asymptomatic, from Hb E “-zero-thalassemia, which is a variably
severe hemolytic anemia.35-37
Screening for “-thalassemias
The red cells of newborns with”-thalassemia
syndromes contain Hb Bart’s, a tetramer of gamma globin that is detected
and reported in most neonatal-screening programs33,34,38,39
(see Table 3). Infants with Hb Bart’s at birth may have a “silent
carrier” state (deletion of one “-globin gene), “-thalassemia trait
(deletion of two genes), or Hb H disease (loss of three genes). Loss of
all four “-globin genes (hydrops fetalis) is incompatible with
intrauterine survival and, thus, is not seen in subjects of newborn
screening. Silent carriers are the largest group with Hb Bart’s at
birth. They have small amounts of Hb Bart’s and do not develop any
clinical or laboratory manifestations. Persons with “-thalassemia trait
generally show a decreased MCV with mild or no anemia. Newborns with
>10% Hb Bart’s by IEF, or >30% Hb Bart’s by HPLC, and/or who develop
more severe anemia may have Hb H disease.33,34,38,39 The
identification of Hb Bart’s in Asian infants can have important genetic
implications for couples, since the cis deletion of both ” genes on a
single chromosome 16 is common in this ethnic group. Thus, couples may
be at risk for subsequent pregnancies complicated by hydrops fetalis.34
All three authors work in Atlanta at Emory
University’s School of Medicine, Department of Pathology and Lab Medicine.
Charbel Abou-Diwan, PhD, is a clinical chemistry post-Doctoral Fellow
there.
Andrew N. Young, MD, PhD, is an associate professor there and medical
director of the clinical laboratory at Grady Hospital.
Ross J. Molinaro, MT(ASCP), PhD, D(ABCC), F(ACB), is an assistant
professor in the department as well as medical director of the core
laboratory at Emory University Hospital Midtown in Atlanta.
References
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an
increasing global health problem. Bull World Health Organ.
2001;79(8):704-712. - Clarke GM, Higgins TN. Laboratory Investigation of
Hemoglobinopathies and Thalassemias: Review and Update. Clin Chem.
2000;46(8):1284-1290. - Vichinsky E. Hemoglobin e syndromes. 2007;79-83.
- Rubin, LP, Hansen K. Testing for hematologic disorders and
complications. Clin Lab Med. 2003; 23(2):317-343. - Steinberg MH. Pathophysiology of sickle-cell disease.
Baillieres Clin Haematol. 1998; 11(1):163-184. - Lane PA. Sickle-cell disease. Pediatr Clin North Am.
1996;43(3):639-664. - Chui DH, Waye JS. Hydrops fetalis caused by alpha-thalassemia: an
emerging health care problem. Blood. 1998;91(7):2213-2222. - Weatherall DJ, Clegg JB. Thalassemia — a global public health
problem. Nat Med. 1996;2(8):847-849. - Colah RB, et al. HPLC studies in hemoglobinopathies.
Indian J Pediatr. 2007;74(7):657-662. - Lafferty JD, et al. The evaluation of various mathematical RBC
indices and their efficacy in discriminating between thalassemic and
non-thalassemic microcytosis. Am J Clin Pathol. 1996;
106(2):201-205. - Hall RBH, Guerra JA, Castleberry CG, et al. Optimizing the detection
of hemoglobin H disease. Lab Med. 1995;26:736-741. - Patrinos GP, Kollia P, Papadakis, MN. Molecular diagnosis of
inherited disorders: lessons from hemoglobinopathies.
Hum Mutat. 2005;26(5):399-412. - Papadea C, Cate JC. Identification and quantification of hemoglobins
A, F, S, and C by automated chromatography. Clin Chem.
1996;42(1):57-63. - Campbell M, Henthorn JS, Davies SC. Evaluation of cation-exchange
HPLC compared with isoelectric focusing for neonatal hemoglobinopathy
screening. Clin Chem. 1999;45(7): 969-975. - Turpeinen U, et al. Two alpha-chain hemoglobin variants, Hb
Broussais and Hb Cemenelum, characterized by cation-exchange HPLC,
isoelectric focusing, and peptide sequencing. Clin Chem.
1995;41(4):532-536. - Cotton F, et al. Evaluation of a capillary electrophoresis method
for routine determination of hemoglobins A2 and F. Clin Chem.
1999;45(2):237-243. - Hempe JM, Craver RD. Quantification of hemoglobin variants by
capillary isoelectric focusing. Clin Chem. 1994;40(12):2288-2295. - Mario N, et al. Capillary isoelectric focusing and high-performance
cation-exchange chromatography compared for qualitative and quantitative
analysis of hemoglobin variants. Clin Chem.
1997;43(11):2137-2142. - Eastman JW, et al. Automated HPLC screening of newborns for
sickle-cell anemia and other hemoglobinopathies. Clin Chem.
1996;42(5):704-710. - Joutovsky A, Hadzi-Nesic J, Nardi MA. HPLC retention time as a
diagnostic tool for hemoglobin variants and hemoglobinopathies: a study
of 60000 samples in a clinical diagnostic laboratory. Clin Chem.
2004;50(10):1736-1747. - Fucharoen S, et al. Prenatal and postnatal diagnoses of thalassemias
and hemoglobinopathies by HPLC. Clin Chem. 1998;44(4):740-748. - Suh DD, Krauss JS, Bures K. Influence of hemoglobin S adducts on
hemoglobin A2 quantification by HPLC. Clin Chem.
1996;42(7):1113-1114. - Shackleton CH, Witkowska HE. Characterizing abnormal hemoglobin by
MS. Anal Chem. 1996; 68(1):29A-33A. - Bergstrome JA, Poon A. Evaluation of a single-tube multiplex
polymerase chain reaction screen for detection of common alpha-thalassemia
genotypes in a clinical laboratory. Am J Clin Pathol. 2002;
118(1):18-24. - Chen YL, et al. Genotyping of alpha-thalassemia deletions using
multiplex polymerase chain reactions and gold nanoparticle-filled
capillary electrophoresis. J Chromatogr A.
2009;1216(7):1206-1212. - Guidelines for the fetal diagnosis of globin gene disorders. Globin
Gene Disorder Working Party of the BCSH General Haematology Task Force.
J Clin Pathol. 1994;47:199-204. - Liu JZ, et al. Detection of alpha-thalassemia in China by using
multiplex ligation-dependent probe amplification.
Hemoglobin. 2008;32(6):561-571. - U.S. Department of Health and Human Services. Agency for Healthcare
Research and Quality. Screening for Sickle-cell disease in Newborns:
Recommendation Statement. U.S.P.S.T.F. (USPSTF) Editor. 2007. - Kaye CI, et al. Newborn screening fact sheets.
Pediatrics. 2006;118(3):
e934-e963. - Consensus conference. Newborn screening for sickle-cell disease and
other hemoglobinopathies. JAMA. 1987;258(9):1205-1209. - Yanni E, et al. Trends in pediatric sickle-cell disease-related
mortality in the United States, 1983-2002. J Pediatr.
2009;154(4):541-545. - Newborn screening: toward a uniform screening panel and system.
Genet Med. 2006;89(suppl): 1:1S-252S. - National Institutes of Health. The management of sickle-cell
disease. L.A.B.I. National Heart. Editor. 2002. NIH Publication
02-2117. - Pass KA, et al. US newborn screening system guidelines II: follow-up
of children, diagnosis, management, and evaluation. Statement of the
Council of Regional Networks for Genetic Services (CORN). J Pediatr.
2000;137(4 Suppl):S1-46. - Krishnamurti L, et al. Coinheritance of alpha-thalassemia-1 and
hemoglobin
E/beta zero-thalassemia: practical implications for neonatal screening
and genetic counseling. J Pediatr. 1998; 132(5):863-865. - Lorey F, Cunningham G. Impact of Asian immigration on thalassemia in
California. Ann N Y Acad Sci. 1998;850:442-445. - Premawardhena A, et al. Hemoglobin E-beta-thalassemia: Progress
report from the International Study Group. Ann N Y Acad Sci.
2005;1054:33-39. - Dumars KW, et al. Practical guide to the diagnosis of thalassemia.
Council of Regional Networks for Genetic Services (CORN).
Am J Med Genet. 1996;62(1):29-37. - Miller ST, et al. A fast hemoglobin variant on newborn screening is
associated with alpha-thalassemia trait. Clin Pediatr. (Phila)
1997;36(2):75-78. - Old JM, Screening and genetic diagnosis of haemoglobin disorders.
Blood Rev. 2003;17(1):43-53.
