Establishing and implementing LDTs utilizing the Test Life Cycle Model
In the United States, there are two types of laboratory tests: U.S. Food & Drug Administration (FDA)-approved or laboratory-developed (LDTs). Most often, FDA-approved tests are marketed by a medical device company and purchased by a laboratory, hospital, or physician’s office. Labs may also develop their own tests in-house: for example, when an FDA-approved test is not available, when an FDA-approved test is modified for a new sample type, or when a new test is more esoteric in nature. The Test Life Cycle Model can be followed by both commercial manufacturers and clinical laboratories to organize the establishment and implementation of either test type.
Definition of an LDT
The FDA considers an LDT to be an in vitro diagnostic (IVD) device that is intended for clinical use and designed, manufactured, and used within a single laboratory.1 These LDTs are sometimes called in-house developed tests, or “home brew” tests.2 The FDA does not consider these devices to be LDTs if they are designed or manufactured completely or partly outside of the laboratory that offers and uses them.1 If a clinical laboratory modifies an FDA-approved test, for example, by changing sample type or intended use, then the test also becomes lab-developed. In the case of an LDT, the lab needs to establish acceptable performance through analytical and clinical validation, and then re-verify performance as part of implementation.
CLIA requirements and limitations
Under the Clinical Laboratory Improvement Act (CLIA), the Centers for Medicare & Medicaid Services (CMS) has regulated laboratories that develop LDTs. CLIA oversees and enforces the accreditation, inspection, and certification of medical laboratories. CLIA requirements address the laboratory’s ability to perform laboratory testing accurately and reliably. CLIA prohibits the release of any test results prior to the laboratory establishing certain performance characteristics relating to analytical validity for the use of that test in the laboratory’s own environment.2
Compliance with CLIA requirements ensures that clinical laboratory practices are of high quality and that the methodologies selected for clinical use have the capability of providing the quality of results required for patient care (42 CFR 493.1445(e)(1) and 42 CFR 493.1445(e)(3)(i–iii)). However, there are no requirements regarding the design, manufacture, and validation of the diagnostic device itself.1
FDA recommendations
Title 21, Section 820 of the Code of Federal Regulations (CFR) sets forth regulations for Quality System Regulation (QSReg) of medical devices and was expected to be applicable to the regulation of LDTs. In late 2014, the FDA issued draft guidance intended for its own staff, clinical laboratories, and the device manufacturer industry for the purposes of FDA notification and reporting for LDTs.1 This guidance was not legally enforceable at the time of its posting, but rather outlined the FDA’s recommendations. While implementation of these regulations for LDTs ultimately never came about, the FDA still has enforcement discretion.
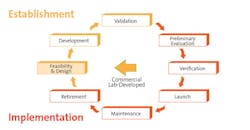
Test Life Cycle Model
Whether a new laboratory test is created by a manufacturer or by clinical laboratorians, there are established measurement evaluations that both follow to ensure the quality and robustness of a laboratory test. The Test Life Cycle Model (Figure 1) can be used to organize the stages of establishment and implementation and the steps taken under both stages of the model needed to assure the development and implementation of a high-quality robust test.3

Establishment stage
The Establishment stage consists of the following steps: feasibility and design, development, and validation.
In feasibility and design, manufacturers or clinical laboratorians can perform a literature review, investigate the clinical usefulness and intended use, perform a feasibility assessment, and assess the legal right to use and perform a marketing assessment. These steps will help decision makers to decide if the appropriate resources should be invested to move the project forward.
The development step encompasses all the iterative steps a manufacturer or clinical laboratorian goes through in coming to a final product or test. Instrumentation is evaluated and chosen for fitness for purpose. Reagents, calibrators, and controls are either purchased or spiked in-house in appropriate matrix. The last steps of development involve the creation of standard operating procedures (SOPs) that will be followed through validation along with determining some preliminary performance characteristics to help again with decisions on whether resources should be invested into moving to the next step/stage. It is important to document each of these steps, conclusions, and decisions made.
In validation, before any method evaluations are performed, a plan should be written to ensure that the proper experiments are planned along with acceptance criteria used to evaluate each experiment/method evaluation. Critical experiments evaluate precision,4 accuracy,5 detection capability,6 analytical specificity,7 stability,8 and clinical validation.9 The measuring10 and reference intervals11 should also be determined. This is not an all-inclusive list; there may be test-, disease state-, or instrument-specific evaluations also. A validation summary should be written to summarize all experiments and determine whether acceptance criteria from the plan were met. The validation summary can also be thought of as a package insert for an FDA-approved test; both give the implementing clinical laboratory specifications to help decide if the test is appropriate for their purpose.
Implementation stage
Whether the assay in question is a commercial FDA-approved kit or an in-house LDT, a clinical laboratory needs to go through the Implementation stage. This stage consists of the following steps: preliminary evaluation, verification, test launch, maintenance, and retirement.
The preliminary evaluation encompasses familiarity and some early performance testing.12 A new instrument, process, or assay may need a period of familiarity before moving further into a clinical laboratory. A clinical laboratory may need to evaluate whether the new kit/instrument fits into the process flow of the lab, and whether the performance criteria outlined from the manufacturer match what is obtained in real life.
Whether the test is purchased from a manufacturer or is an LDT, the implementing lab will need to perform verification to ensure that the test meets acceptance criteria set for the new test. Verification can be thought of as a small validation experiment, encompassing some of the same experiments but with smaller numbers over a shorter time span. A verification plan with pertinent experiments and acceptance criteria should be written. Precision, accuracy, and detection capability should be evaluated along with verifying the measuring and reference interval.
If verification is completed and experiments meet acceptance criteria, then a new test can be launched into the clinical laboratory.13 A clinical laboratory will still perform test maintenance during the lifetime of the test. Finally a test may be retired due to implementation of a new version.
Meeting the standard
The goal of both FDA-approved test manufacturers and clinical laboratories that develop LDTs is to produce a test that meets a standard of high quality. Established measurement procedures are followed by creators of LDTs, whether the creator is a manufacturer or a clinical laboratorian. The Test Life Cycle Model organizes the establishment and implementation of a new or modified test and can be used to develop and implement a high quality, robust test.
REFERENCES
- U.S. Food and Drug Administration. Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories: FDA Notification and Medical Device Reporting for Laboratory-developed Tests (LDTs). https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm416685.pdf.
- Centers for Medicare & Medicaid Services. CLIA overview: laboratory-developed tests (LDTs) frequently asked questions. https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Downloads/LDT-and-CLIA_FAQs.pdf.
- CLSI. A Framework for Using CLSI Documents to Evaluate Clinical Laboratory Measurement Procedures. 2nd ed. CLSI report EP19. Wayne, PA: Clinical and Laboratory Standards Institute; 2015.
- CLSI. Evaluation of Precision of Quantitative Measurement Procedures. CLSI document EP05. Wayne, PA: Clinical and Laboratory Standards Institute; 2014.
- CLSI. Measurement Procedure Comparison and Bias Estimation Using Patient Samples; Approved Guideline—Third Edition. CLSI document EP09-A3. Wayne, PA: Clinical and Laboratory Standards Institute; 2013.
- CLSI. Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures; Approved Guideline—Second Edition. CLSI document EP17-A2. Wayne, PA: Clinical and Laboratory Standards.
- CLSI. Interference Testing in Clinical Chemistry; Approved Guideline—Second Edition. CLSI document EP07-A2. Wayne, PA: Clinical and Laboratory Standards Institute; 2005.
- CLSI. Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline. CLSI document EP25-A. Wayne, PA: Clinical and Laboratory Standards Institute; 2009.
- CLSI. Assessment of the Diagnostic Accuracy of Laboratory Tests Using Receiver Operating Characteristic Curves; Approved Guideline—Second Edition. CLSI document EP24-A2. Wayne, PA: Clinical and Laboratory Standards Institute; 2011.
- CLSI. Evaluation of the Linearity of Quantitative Measurement Procedures: A Statistical Approach; Approved Guideline. CLSI document EP06-A. Wayne, PA: Clinical and Laboratory Standards Institute; 2003.
- CLSI. Defining, Establishing, and Verifying Reference Intervals in the Clinical Laboratory; Approved Guideline—Third Edition. CLSI document EP28-A3c. Wayne, PA: Clinical and Laboratory Standards Institute; 2008.
- CLSI. Preliminary Evaluation of Quantitative Clinical Laboratory Measurement Procedures; Approved Guideline—Third Edition. CLSI document EP10-A3-AMD. Wayne, PA: Clinical and Laboratory Standards Institute; 2014.
- CLSI. User Protocol for Evaluation of Qualitative Test Performance; Approved Guideline—Second Edition. CLSI document EP12-A2. Wayne, PA: Clinical and Laboratory Standards Institute; 2008.
Paula Ladwig, MS, MT(ASCP), serves as a development technologist coordinator for Mayo’s Clinical Mass Spectrometry Development Laboratory, where her focus is the quantitation of proteins. She holds a Master’s Degree in Biochemistry and Structural Biology through the Mayo Graduate School.
