SNP genotyping with the next generation of CGH microarray

Although genetic variation is a major driving force behind evolution, certain variants also underlie many congenital diseases. Copy number variants (CNVs) are a major source of such genetic variation, and are defined as chromosomal segments at least one thousand bases in length that vary in copy number between individuals. The prevalence of CNVs throughout the general population suggests that they represent a significant proportion of total genomic variation, and it has been estimated that CNVs may affect as much as 4% to 5% of the human genome.1 However, CNVs occurring within coding or regulatory regions of the genome can also have an adverse effect on gene expression.
A second contributor of variation is single nucleotide polymorphism (SNP), in which two distinct alleles are possible at a single genome position, with each allele appearing at high frequency within the population. SNP genotyping has many applications in medical research, such as providing information on the level of heterozygosity between two chromosomes. This is particularly important in the context of genetic disease, since runs of homozygosity (ROH) increase the incidence of recessive mutations being expressed as a disease phenotype.
Analysis of CNVs and SNPs is therefore an important part of constitutional genetics research in children displaying signs of developmental disorders. Although information on both types of genetic variation is complementary in many of these investigations, each has traditionally been analyzed using separate approaches.
The gold standard for CNV detection is array comparative genomic hybridization (aCGH) using long oligonucleotide microarray platforms. This well-established technique benefits from fast processing and minimal hands-on time. In contrast to this efficient approach, SNP analysis is usually accomplished using high-resolution SNP microarrays. With high resolution comes increased data complexity, requiring extra resources of time and money to unravel this information and highlight biologically relevant insights. The reality of the clinical research laboratory instead demands a certain level of pragmatism, and a balance must be met between the size of the aberrations reported and the overall efficiency of a given approach.
Streamlining both CNV and SNP analysis of the same sample, microarray platforms are now available that incorporate SNP probes onto the aCGH array. Designed specifically for molecular cytogenetics research, these new arrays enable the effortless integration of SNP analysis into existing aCGH workflows, providing additional insight into underlying disease mechanisms, at little extra cost. Reflecting the needs of clinical research laboratories, such arrays combine high numbers of CNV probes, enabling highly accurate CNV detection, with sufficient SNP probes to reliably detect larger ROH. Interestingly, for reasons that will be later discussed, only larger ROH are likely to underlie disease, which is important when considering the role of the combined CNV and SNP array in the modern laboratory.
aCGH – the gold standard for CNV studies
Within the field of cytogenetics, molecular approaches are becoming ever more important in identifying the genetic factors contributing to a disease or syndrome. This is reflected in the widespread use of the aCGH platform, a molecular cytogenetic approach for detecting and locating any net gains or losses of genetic material.
In essence, aberrations within the test genome are detected through comparison with a normal reference genome. DNA fragments from a normal reference sample and a test sample are first labeled with two different fluorophores. Equal quantities of the two samples are then mixed, and competitively co-hybridized to a DNA microarray of several thousand evenly spaced cloned oligonucleotides.
If a single genomic region has an equal DNA content in the test and the normal reference sample (reflecting a normal CNV profile), the corresponding oligonucleotide on the microarray surface will emit an equal intensity of both fluorophores. On the other hand, if the test sample contains a CNV in a particular genomic region, this loss or gain of DNA is revealed as a shift in the fluorescence ratio.
Array CGH has proven to be a specific, sensitive, and fast technique, amenable to automation for high-throughput workflows. Because the exact genomic position is known, aberrations can also be mapped directly onto the chromosomal location. Well established in many laboratories, new technologies are rapidly expanding the applications possible with the aCGH platform.
The next generation of aCGH
Within constitutional genetics research, SNP genotyping has three main applications:
- Identification of uniparental disomy (UPD), through detection of ROH;
- Identification of ROH by inheritance, due to descent or consanguinity;
- Aiding in the identification of mosaic aneuploidy and chimerism.
In these three cases, the combination of SNP and CNV data provides unique insight into the underlying genetic causes of disease phenotypes, as approximately 80% of developmental disorders of unknown cause have a normal result from traditional aCGH analysis. aCGH can only detect net gains or losses in genomic content, missing copy-neutral aberrations such as ROH. While SNP analysis can detect these latter features, SNPs do not occur throughout the whole genome, and SNP analysis may fail to detect some CNVs. In order to identify both CNVs and copy-neutral aberrations, traditional workflows would require the use of two separate arrays, but high-resolution SNP microarrays are time-consuming and labor-intensive: a standard SNP microarray protocol can take up to four days to complete, including around seven hours of hands-on time (Figure 1).
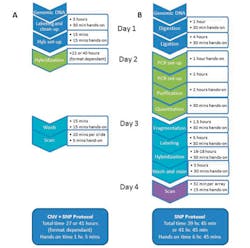
Figure 1. A comparison of two typical array processing workflows. The combined CNV + SNP workflow offers considerable time savings when compared to a typical SNP genotyping platform. A: An example protocol for the combined CNV + SNP array; B: A typical protocol for an SNP genotyping platform. (Courtesy of Oxford Gene Technology.)
Improving the efficiency of these studies, recent advances in probe design have now made it possible to incorporate SNP probes onto the aCGH platform, allowing the simultaneous analysis of CNVs and SNPs on a single aCGH and SNP array. For example, Figure 2 shows how the combined aCGH and SNP array enables the analysis of both CNV and ROH data in a consanguineous sample. The main challenge in developing this approach lies in selecting SNP probes that reliably detect and discriminate between SNP alleles while working under hybridization conditions optimized for CNV detection. Choosing the most suitable and insightful SNP probes therefore requires much careful optimization, and several companies now provide these array platforms.

Figure 2. CNV and ROH are both easily identified on a consanguineous sample, using the combined CNV + SNP array with supporting software. The total percentage of homozygosity is indicated above the ideograms, reflecting the genotype of SNP probes shown to the left, where black indicates a heterozygous genotype, and red indicates homozygous. CNV data is displayed by solid blocks beside the ideogram, with deletion shown as red blocks, and amplifications as green blocks (data provided by Emory Genetics Laboratory).
The combined array delivers high quality CNV data2 and provides a more streamlined workflow compared to SNP-based array platforms (Figure 1). This is particularly useful for high-throughput research laboratories, allowing the integration of SNP analysis into existing aCGH workflows.
Analysis of UPD with the combined array
The main application of SNP analysis in cytogenetic research is for the detection of ROH, which is often linked to disease incidence via homozygous mutant alleles. Although smaller regions of ROH can arise due to consanguinity or shared parental ancestry,3 the most common cause of ROH is uniparental disomy, occurring when both copies of a chromosome are inherited from a single parent. The inheritance of two copies of an identical chromosome is known as isodisomy, and results in whole chromosome ROH. Known as heteroisodisomy, inheritance of two different chromosomes from the same parent is also possible, but since these chromosomes remain heterozygous, this is unlikely to cause a disease phenotype. It is estimated that the frequency of UPD in newborns is approximately 1 in 3,500 with not all UPDs causing a phenotypic effect, and around 1,100 cases of whole chromosome UPD have been described in the literature.4
Because UPD is a copy-neutral aberration, it cannot be detected using a traditional aCGH, and a platform containing SNP probes must instead be used. Interestingly, because UPD is often associated with chromosomal aberrations, it is important to study UPD using a combined CNV and SNP platform. In a third of clinical cases, UPD is actually uncovered through a chromosomal aberration.
ROH through descent and consanguinity
In clinical genetics, consanguinity is defined as the union of individuals related as second cousins or closer, and it is thought that such couples account for 10.4% of the world’s population.5 Consanguinity samples have a significantly increased number and size of ROH exceeding 10 Mb,6 and it is estimated that the offspring of first cousins have a 1.7% to 2.8% greater risk of congenital malformations, in comparison with the outbred population.
The number and size of ROH in offspring of consanguineous unions depends on the degree of parental relatedness,6,7 and this introduces an important factor in terms of identifying biologically relevant ROH that are actually linked to disease incidence. This remains a challenge, since ROH also occurs in outbred populations (termed ancestral ROH). This has been well studied, and in normal European populations, ROH covering on average 93 Mb of DNA were present throughout the genome. These ROH can be up to 4 Mb in length8 and were found in populations from all parts of Europe, with a healthy individual carrying an average of 40 regions of ROH, with a median length of approximately 1.25 Mb.9 A large study of diverse populations showed that many contain relatively short ROH,3 and ROH of over 10 Mb is considered very rare in cosmopolitan populations. All ROH have the potential to cause an autosomal recessive disease. However, it is the excessively long ROH that increase the chance of a discernible phenotype, and so the resolution must be correctly set in order to exclude benign ancestral ROH.
Biological relevance of ROH: choosing the correct SNP resolution
In SNP detection, a higher resolution enables the detection of smaller ROH. However, detecting smaller ROH is of little biological relevance, and this is a crucial factor to consider in the application of the combined aCGH and SNP platform. As such, combined aCGH and SNP platforms focus on delivering the high-resolution CNV content required, while also providing sufficient SNP probes for accurate detection of ROH at a resolution of approximately 7 Mb. While this resolution is not as high as dedicated high-resolution SNP arrays, it reflects the reporting requirements of clinical research laboratories that do not want to detect underlying ROH (i.e., ancestral ROH). A lower-resolution format is also more cost-effective, allowing more samples to be run in parallel.
There is conflicting evidence in the literature regarding the resolution cutoff value that should be used, summarized in Figure 3. The variation in cutoff values reported in the literature is reflected in the different reporting policy of many research laboratories. A recent study found that each laboratory made its own decision regarding the cutoff value for classifying biologically relevant ROH, ranging from ³10 Mb to ³5 Mb.10 In some laboratories the total percentage of homozygosity across the genome was reported, whereas in other laboratories the frequency of ROH was considered to be important. Overall there was much variability in what was considered biologically relevant, highlighting the need for the introduction of guidelines to standardize the process.

Figure 3. Several recent studies present conflicting recommendations regarding the cutoff value that should be used to distinguish ancestral ROH from biologically relevant ROH. (Courtesy of Oxford Gene Technology.)
Understanding complex cytogenetic disorders
The combined aCGH and SNP platform is also ideal for the analysis of complex chromosomal conditions such as mosaicism and chimerism, which are characterized by the existence of multiple cell lines in the same individual carrying distinct genotypes. While mosaicism arises from meiotic or mitotic errors early in development, chimerism instead results from the fusion of multiple fertilized zygotes, and these conditions have been reported for a number of chromosomal aberrations, from aneuploidy to CNVs.11
Although mosaic aneuploidy (the most common form of mosaicism) can be detected using standard aCGH, SNP data also provides additional data confirmation. Furthermore, the percentage of cells carrying the aberration can be determined. This is calculated from the B-allele frequency (BAF) of each SNP probe, which is a normalized measure of the allelic intensity ratio of two alleles (A and B) throughout a population. For example, a BAF of 1 or 0 indicates the complete absence of one of the two alleles (e.g., AA or BB), and a BAF of 0.5 indicates the equal presence of both alleles (e.g., AB). This, therefore, provides a measure of an SNP genotype across the cell population, reflecting distinct SNP genotypes arising from different cell populations.
Chimerism was not often detected prior to the introduction of SNP genotyping, as the genotypes of multiple cells would be compared only if it was suspected from an individual’s phenotype. Genome-wide SNP analysis instead enables the detection of distinct genotypes belonging to different cell populations, as part of an initial study using the combined aCGH and SNP array.
Summary
Enabling the simultaneous detection of CNV and copy-neutral aberrations within one test, the combined aCGH and SNP array has the potential to vastly improve both workflow efficiency and productivity within the cytogenetics research laboratory. The applications of the combined array are summarized in Figure 4.
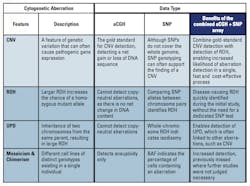
Figure 4. Summarizing the application and benefits afforded by simultaneous CNV and SNP data acquisition on the combined aCGH and SNP array platform. Incorporating SNP analysis into the aCGH array provides additional insight into underlying genetic conditions. (Courtesy of Oxford Gene Technology.)
Standard aCGH is a rapid, sensitive, and high-throughput approach for CNV detection, and although central to many cytogenetic studies, approximately 80% of developmental disorder samples yield a normal result, highlighting the need for additional information. Incorporating SNP analysis into the aCGH array provides insight into the underlying genetic causes, while retaining the benefits of the well-established aCGH platform. In many cases this removes the need for time-consuming follow-up studies, and is particularly beneficial considering the costs and labor involved in a high-resolution SNP array.
Due to the prevalence of ancestral ROH, it is clear that there is little additional benefit to analyzing ROH at high resolution, which adds to the complexity of the data, increasing the laboratory workload. The most effective combined aCGH and SNP arrays instead provide sufficient resolution to detect abnormally long ROH stretches present in UPD or consanguineous samples, while excluding standard length ancestral ROH that are not biologically relevant, without compromising CNV detection.
This new generation of combined aCGH and SNP array presents a high-throughput and efficient alternative to high-resolution SNP platforms, incorporating SNP analysis into existing aCGH workflows, and yielding unique biological insights with minimal additional cost.

References
- DF Conrad, Pinto D, Redon R, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704-712.
- Curtis C, Lynch AG, Dunning MJ, et al. The pitfalls of platform comparison: DNA copy number array technologies assessed. BMC Genomics. 2009;10:588-610.
- Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF. Genomic runs of homozygosity record population history and consanguinity. PLoS One. 2010;5(11):e13996.
- Liehr T. Cytogenetic contribution to uniparental disomy (UPD). Mol Cytogenet. 2010;3:8.
- Bittles AH, Black ML. Consanguinity, human evolution, and complex diseases. Proceedings of the National Academy of Sciences USA. 2010;107:1779-1786.
- Sund KL, Zimmerman SL, Thomas C, et al. Regions of homozygosity identified by SNP microarray analysis aid in the diagnosis of autosomal recessive disease and incidentally detect parental blood relationships. Genet Med. 2013;15(1):70-78.
- Bennett RL, Motulsky AG, Bittles A, et al. Genetic counseling and screening of consanguineous couples and their offspring: recommendations of the National Society of Genetic Counselors. J Genet Counseling. 2002;11(2):97-119.
- McQuillan R, Leutenegger AL, Abdel-Rahman R, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83;(3)359-372.
- Nothnagel M, Lu TT, Kayser M, Krawczak M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum Mol Genet. 2010;19(15):2927-2935.
- Grote L, Myers M, Lovell A, Saal H, Lipscomb Sund K. Variability in laboratory reporting practices for regions of homozygosity indicating parental relatedness as identified by SNP microarray testing. Genet Med. 2012;14(1):971-976.
- Hassold TJ, Jacobs PA. Trisomy in man. Annu Rev Genet. 1984;18:69-97.
- Kearney HM, Kearney JB, Conlin LK. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med. 2011;31(4):595-613.
- Conlin LK, Thiel BD, Bonnemann CG, et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010;19(7):1263-1275.
- Papenhausen P, Schwartz S, Risheg H, et al. UPD detection using homozygosity profiling with a SNP genotyping microarray; part A. Am J Med Genet. 2011;155A(4):757-768.
- Bruno DL, Ganesamoorthy D, Schoumans J, et al. Detection of cryptic pathogenic copy number variations and constitutional loss of heterozygosity using high resolution SNP microarray analysis in 117 patients referred for cytogenetic analysis and impact on clinical practice. J Med Genet. 2009;46(2):123-131.
