The proper handling of CJD-infected patient samples in the pathology laboratory

The term prion was introduced in 1982 by neurologist Stanley B. Prusiner1 to describe a proteinaceous infectious particle that was associated with the disease scrapie. Prions are pathologic infectious agents composed of bits of misfolded protein that cause other proteins to fold in multiple, structurally abstract ways. Prion diseases are rare neurodegenerative disorders identified as transmissible spongiform enchephalopathies (TSEs).
History of prion diseases
Several hundred years ago, sheep herders noted that some of their sheep became ill, appeared to be deranged, and started scraping their hindquarters. This sheep disease became known as scrapie. For many years this disease was not well understood, with the causative agent remaining elusive. Unlike most bacteria and viruses, prion protein (PrP) is hearty and is not susceptible to high temperatures and many common disinfectants.2
In the mid-20th century, while scientists were studying kuru, another neurological disease found in the people of Papua New Guinea, it was shown that this disease could be transmitted to chimpanzees, as could a third neurological disease, Creutzfeldt-Jakob Disease (CJD). Further studies showed that scrapie, kuru, and CJD all acted in similar ways, creating severe neurologic abnormalities within the brain. Prusiner demonstrated that this (scrapie) agent was not a bacteria or virus because it lacked DNA and RNA, but was actually a protein.3,4
Prion protein (PrP)
Over the years, other prion diseases have been identified in both humans and animals (Table 1). In humans, cases of CJD and kuru (attributed to cannibalistic practices) have been seen. While scrapies was well known, “mad cow disease” (bovine spongiform encephalopathy) became relevant when individuals contracted a form of it (known as variant CJD, or vCJD) when ingesting contaminated beef.2
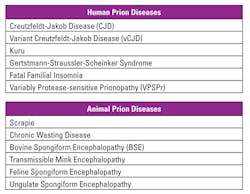
Studies have shown that the prion protein can be found in healthy individuals due to a gene found on chromosome 20(p13).5 This prion protein gene (PRNP) in its healthy state causes no symptoms or disease. However, it can misfold and can further “teach” other proteins to misfold.6 These misfolded proteins lead to TSEs.7
In the United States, about 300 cases of TSEs are reported each year and have been associated with fatal degenerative brain diseases. TSEs are characterized by microscopic vacuoles and amyloid deposition in the brain, similar to those deposits seen in Alzheimer’s disease and Parkinson’s disease.2
TSEs: types and symptoms
There are three types of TSEs seen in humans. The infectious form (less than one percent incidence) is spread by consuming infected foods, iatrogenic spread of infected tissues (organ transplant, contaminated human growth hormone, etc.), and transfusion. The sporadic form is the most common and is responsible for 85 percent to 90 percent of all classic CJD cases. One or two people out of a million die every year from sporadic CJD, which appears later in life, between the ages of 55 and 75. The familial form occurs due to an autosomal, dominant gene mutation of PrP. About 10 percent to 15 percent of the total human TSEs are inherited. Fatal familial insomnia interferes with sleep and eventually leads to the deterioration of mental and motor brain functions.8,9
Symptoms of TSEs are progressive and can include headaches, joint pain, difficulty in walking/talking, seizures, paralysis, and loss of bodily and cognitive functions. These neurodegenerative disorders have a long incubation period, estimated to be about 11 years up to 40 years, depending on the type.10 Various treatment modalities have been tried with no success, as death is always imminent once symptoms appear.
Guidelines for handling known or suspected tissues/fluids from patients with or at risk for CJD/vCJD
Infectivity is most often found in the central nervous system, specifically the brain, spinal cord, and eye. Table 2 identifies tissues that are proven or assumed to have medium to high infectivity for TSEs. Dura mater/anterior eye/cornea are considered high-risk tissue sources even though PrPTSE (disease-associated form of the prion protein) may not be detected.
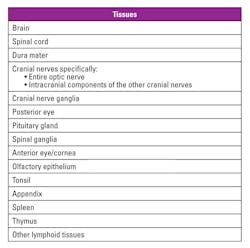
Peripheral tissues have been found to carry low levels of prion protein and are considered low-risk. Cerebrospinal fluid (CSF) and blood are also classified as having low-risk infectivity. However, vCJD has been transmitted via contaminated blood transfusions. The most recent TSE described is variably protease-sensitive prionopathy (VPSPr).11
Decontamination of infected materials12,13
Agents of TSEs are generally resistant to routine disinfectants as well as common sterilizing methods used in the laboratory. Ineffective disinfectants include alcohol, ammonia, chlorine dioxide, formalin, glutaraldehyde, hydrogen peroxide, and iodine. Ineffective sterilization processes include dry heat, UV light, microwaving, autoclaving (at 121oC for 15 minutes), glass bead sterilizers, and certain gases (ethylene oxide or formaldehyde). Effective disinfectants include 2M NaOH for one hour.
Precautions in handling infectious materials
Frozen section examination should not be performed on high-risk tissues for patients with or at risk for CJD or vCJD. Gross examination of tissues requires following routine universal precautions for blood-borne pathogens and wearing appropriate personal protective equipment (PPE). Use of cut-resistant steel mesh gloves and disposable instruments is strongly advised. Gross examination can be performed on fresh or fixed tissue, preferably in a class I biological safety cabinet, using disposable paper lining over the base of the cabinet to absorb contaminated splashes.
Surgical instruments and the grossing area can be decontaminated with 2M NaOH for a minimum of one hour. Disposable instruments and sharps can be decontaminated with 2M NaOH for one hour prior to placing them in a suitable biohazard container. All waste, including gowns, gloves, and aprons, should be incinerated. Non-disposable instruments and cut resistant/steel gloves should be decontaminated with 2M NaOH for one hour.
Any contaminated glassware, including microscope slides, should be collected in a suitable container, double-bagged, and incinerated. Work surfaces, including microtome for non-formic treated blocks and biohazard safety cabinet, should be washed with 2M NaOH. Any contaminated fluids, fixatives, or solvents can be absorbed in sawdust in a combustible, stout container with lid, double-bagged, and incinerated. Blood and CSF samples can also be handled in a similar manner.
Tissue samples can be fixed in 10 percent neutral buffered formalin for 24 hours followed by a one-hour fixation in 96 percent formic acid. Tissues can then be removed, washed thoroughly with tap water, and routinely processed like other pathology specimens. Fluids used to process tissue samples can be disposed of as per routine protocols/precaution procedures. Microtomy can be performed using disposable blades as per routine protocol.
Retrospective diagnoses and special circumstances
In cases where a retrospective diagnosis of CJD after normal tissue sample processing has occurred, follow-up is required. Take into account the following when considering what action to take:
- The type of tissue(s) involved.
- The type of CJD that has been diagnosed and how recently the diagnosis was made.
- Whether the patient had clinical symptoms of CJD or was in one of the “at risk” groups.
- Whether the case was reported to the National
- Prion Disease Pathology Surveillance Center (NPDPSC) and whether any of the tissue should be referred for further special investigations.
The following are action steps under these circumstances:
- Any samples taken from the patient should be traced, including stored specimens.
- Fixed tissue should be double-bagged and sent for incineration after the statutory six weeks.
- Any residual frozen tissue, including cryostat sections, should be fixed per CJD protocol, double-bagged, and sent for incineration.
- The paraffin blocks should be removed from main store and filed separately, clearly labeled as form of CJD, and then stored separately. Reprocessing the blocks using formic acid is not required.
- Unmounted slides should be sent for incineration. Mounted (cover-slipped) slides can be filed as per routine protocol.
- Prior to incineration, the NCJDSC should be contacted to ascertain whether it has a use for fixed/frozen tissue(s).
- If it is still in use, the microtome blade used to cut the sample should be placed in a sharps box and disposed of by incineration. The water bath should be changed and cleaned with 2M NaOH for 60 minutes.
- For brain banking, identify the brass plates used for dissection of the CJD sample. They can no longer be used for the dissection or freezing of brain tissues. Prions are known to bind to metal surfaces, and washing with water will not guarantee the removal of prion infectivity. Frozen tissue from the next two cases received and cut fresh on the same brass plate following the CJD sample should be identified and withdrawn from research and either stored with appropriate biohazard labeling or transferred to NPDPSC.
Here’s where to send tissue samples for CJD testing: National Prion Disease Pathology Surveillance Center; Institute for Pathology; Case Western Reserve University; Cleveland, OH 44106. Email: [email protected].
Occupational exposure guidelines
Based on the World Health Organization (WHO) “Infection Control Guidelines for Transmissible Spongiform Encephalopathies (TSE or Creutzfeldt-Jacob Disease, CJD)” and the information received from Centers for Disease Control and Prevention (CDC), Prion and Public Health Office, there have been no confirmed cases of occupational transmission of TSEs to humans, but it is prudent to take a precautionary approach.
The highest potential risk from exposure to high or low infectivity tissues is through direct inoculation (for example, needle-sticks, puncture wounds, sharps, injuries, or contamination of broken skin). Exposure by splashing or unintentional ingestion may be considered a hypothetical risk and should be avoided.
CSF and blood are classified as low-risk tissues and do not require special precautions for biochemical or cytological testing. Regular precautions applied in the laboratory by following proper laboratory safety procedures and use of PPE for any blood or body fluid sample are protective against CJD as well.
CJD and other forms of TSEs are rare in occurrence. However, the presence of prion disease cannot be taken lightly. Due to the lengthy incubation period, it becomes an extreme hazard that requires every effort to quickly identify. Protocols for handling and processing contaminated specimens must be part of every laboratory manual. Treatment for prion disease is mainly supportive and no cure is available at this time.
REFERENCES
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie.
Science. 1982;216(4542):136-144. - Prion Alliance. What are prions? Nov 26, 2013. http://www.prionalliance.org/faq.
- Gajdusek DC. Experimental transmission of a Kuru-like syndrome to
chimpanzees. Nature. 1966;209(5025):794-796. - Prusiner SB. The prion diseases. Brain Path. 1998;8(3):499-512.
- Oesch B, Westway D, Walchli M, et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell. 1985;40(4):735-746.
- Jucker M, Walker LC. Self-propagation of pathologic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45-51.
- Kocisk DA, Come JH, Priola SA, et al. Nature. 1994;370(6489):471-474.
- Klug GM, Wand H, Simpson M, et al. Intensity of human prion disease
surveillance predicts observed disease incidence. J Neurol Neurosurg Psych. 2013;84(12):1372-1377. - Appleby BS, Lyketsos CG. Rapidly progressive dementias and the treatment of human prion diseases. Expert Opin Pharmacother. 2011;12(1):1-12.
- Krause EK, Singh NN. Variant Creutzfeldt-Jakob Disease and bovine spongiform encephalopathy clinical presentation. Medscape. Jan 11, 2016. https://emedicine.medscape.com/article/1169688-clinical.
- Budka H, Aguzzi A, Brown P, et al. Tissue handling in suspected Creutzfeldt-Jakob disease( CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathology. 1995;5(3):319-322.
- Rutala WA, Weber DJ. Guideline for disinfection and sterilization of prion-contaminated medical instruments. Infection Control and Hospital Epidemiology. 2010;31(2):107-117.
- WHO infection control guidelines for transmissible spongiform encephalopathies. Report of a WHO consultation. Geneva, Switzerland, 23-26 March 1999. www.cdc.gov/prions/cjd/infection-control.html.
Nadine Pizzella, MT(ASCP), serves as the Anatomic Pathology Manager for NorDx Laboratories, Scarborough, ME.
Anthony Kurec, MS, H(ASCP)DLM, is Clinical Associate Professor, Emeritus, SUNY Upstate Medical University, Syracuse, NY, and a member of MLO’s Editorial Advisory Board.
