A holistic overview of hemoglobinopathies interactions with glycated hemoglobin
The laboratory medical director and technical staff are dedicated to providing accurate hemoglobin A1C (HbA1c) test results that are free from endogenous interferences such as hemoglobinopathies (inherited blood disorders and diseases that primarily affect red blood cells). It is important to remember that in the world of hemoglobinopathies, there are myriads of combinations that could potentially interact with A1c (also known as glycated hemoglobin) and impact not only the measurement of HbA1c, but also the in vivo production of HbA1c itself. Ultimately, identifying the clinical status of the patient, will lead to more accurate result interpretation and better patient treatment decisions.
The interference of hemoglobin variants or elevated hemoglobin F (HbF) in the determination of A1c has been discussed in different studies, often with a limited number of selected cases and always with a look at hemoglobinopathies separately.1,2,3,4 Lacking in the discussion is consideration of hemoglobin variants that are coinherited with thalassemia and/or in association to elevated HbF. Common and less common genetic interactions should also be considered when evaluating their possible interference on A1c. The magnitude of the interference on A1c of a specific hemoglobin variant, in a defined state (heterozygous, homozygous, combined heterozygous), cannot be fully evaluated without considering these interactions in all their concurrent elements.
The most common interactions between Hb variants, elevated HbF, and thalassemia include the following:
- HbS (sickle hemoglobin) is often coinherited with alpha thalassemia (30% of African American carry also alpha thalassemia).5,6
- HbS Arab/Indian haplotype is typically with raised HbF.7
- HbE, HbC, HbD are often associated with high HbF.
- HbE is a thalassemic variant.8
- Common hemoglobin variants (HbS, HbE, HbC, HbD) are associated with beta and alpha thalassemia.9
- Common hemoglobin variants (HbS, HbE, HbC, HbD) are associated with a double heterozygosity form.10
- Raised HbF can be due to the clinically silent hereditary persistence of fetal hemoglobin (HPFH).11
- Raised HbF can be due to delta beta thalassemia and beta thalassemia, which affect the red blood cells (RBCs).12,13,14,15
- Rare Hb variants can be in association with any of the above.16
All of these combinations can interfere with the formation of A1c for a number of different reasons. First, the presence of a variant globin chains tetramer could alter the normal interaction of glucose with hemoglobin A (HbA).17 Second, the hematological changes caused on RBCs could influence the journey of the glucose across the membrane before its interaction with HbA, to make A1c.18 Third, these combinations could affect the measurement of A1c, depending on the testing method used.
When studying the possible mechanisms that could interfere with A1c testing, it is important to consider the high level of complexity and interactions due to coinherited hemoglobinopathies. For instance, if an individual carries a variant and elevated HbF, the interference on HbA1c of the former must be considered in combination with the interference of the latter.
A1c – Hb glycation and interfering conditions
It is widely accepted that hemoglobin A1c is equivalent to mean blood glucose (MBG), and it is used routinely to monitor blood glucose control in diabetes.19 The formation of A1c is the result of the glycation process that is subject to a series of kinetic mechanisms and dynamics that should be studied with a holistic approach.20 At the molecular level, a molecule of plasma glucose must cross the RBC membrane and reach the N-terminal valine of the beta chain of hemoglobin A. It reacts reversibly first (Schiff base) and then stabilizes to form A1c.
HbA1c depends on the rate at which hemoglobin is glycated and the time frame of glycation. The rate is dependent on MBG and on the kinetics of the reaction of glucose at the N-terminal valine of the beta normal chain. The glycation time is 120 days, the average circulating lifespan of RBCs from the production in the bone marrow to their removal in the spleen and liver.
The physiological variation in size, volume, and hemoglobin content of the RBCs from nascence (reticulocytes) through senescence are factors that can influence the glycation process. The RBC’s variation in size distribution is measured by RDW (red blood cell distribution width). Blood HbA1c is the mean for RBCs with values that range from very low in the reticulocytes to approximately twice the mean in the oldest RBCs.21 An increase of RDW or an increase in reticulocytes implies a variation in the size and permeability of the RBC membrane, which could influence glycation.
Clinical conditions and hemoglobinopathies that alter the RBC lifespan and the homeostatic mechanisms that control the ratio between red cell production in the bone marrow and destruction in the spleen and liver could also affect glycation.
Hb variants
When referring to glycation, hemoglobin variants in the heterozygote condition (AS, AE, AC, etc.) are generally considered like the wild type of AA.22 Though, some differences have been seen because the normal beta chains are not the same as altered beta (S,C,D,E). Studies have demonstrated for instance that individuals with an AS trait have a median A1c value 5% higher than those with AA.23 Moreover, knowing how common the prevalence of a compound heterozygosity AS with alpha or beta thalassemia, as well as an AS trait with elevated HbF typical in the Arab/Indian haplotype, we then understand the risk of reporting A1c with an incomplete hematological profile necessary for a thoughtful evaluation of glucose control in the patient.7
The risk is even higher if we report as “A1c” the glycated fractions (S1c, C1c, etc.) of individuals with homozygous or double heterozygous conditions (SS, CC, SC) where there is no HbA.24 Thus, there is no A1c. In these cases, the rate of glycation is not like that in a normal phenotype AA, because these conditions of homozygous variants have decreased red cell survival. The reported A1c would, in fact, be an inaccurate surrogate.25
Thalassemia
When classifying the hematological presentation and clinical features of hemoglobinopathies, we tend to forcibly divide between heterozygote and homozygote conditions, as if there was a categorical barrier between the two. Recent studies have looked at the thalassemia syndromes as a unique disease with different grades of phenotype severity from the beta thalassemia trait to thalassemia intermedia up to thalassemia major.26 In these conditions (trait, intermedia, major), decreasing amounts of HbA are counterbalanced by an increased level of HbF, up to more than 90% in the homozygote condition.
In beta thalassemia, the main pathophysiological mechanism is due to the precipitation of unbalanced alpha chains that could cause damage to the RBC membrane, which would result in typical RBC morphologic changes in the RBCs such as microcytic RBCs, increased number of RBCs, variability in their size (anisocytosis), and different levels of anemia. A certain degree of ineffective erythropoiesis (altered production of RBCs) is also seen in beta thalassemia carriers and a reduction in red cell survival could have an impact on glycation and HbA1c formation.27
These alterations are obviously less severe in carriers than in affected patients. However, in some individuals, it could be different. Heterozygous β-thalassemia could lead to the phenotype of thalassemia intermedia instead of the asymptomatic carrier state.13 In alpha thalassemia, the excess of unmatched beta globin chains has been proven to have specific effects on ineffective erythropoiesis, hemolysis, targeted oxidant attack, state of cell hydration, membrane mechanical stability, and deformability.28,29 These alterations on the RBC membrane, due to alpha thalassemia, could impact the glucose crossing of the membrane and ultimately the glycation process.
HbH disease, which is the most severe form of alpha thalassemia, causes hemolysis and a shortened RBC life span, which could produce a misleadingly low A1c result.30 Alpha thalassemia is often co-inherited with HbS and HbE. Both conditions (variant and thalassemia) converge in causing an alteration of the glycation process. For this reason, it is important to be aware of the presence of any variants, as well as thalassemia and HbH in particular, due to the risks of their combined effects on the RBC physiology and life span.
Hemoglobin F
Fetal hemoglobin (HbF) in adults is typically below 1%. Raised levels of HbF in adults can be due to different conditions: pregnancy, aplastic anemia, drugs, thalassemia, delta beta thalassemia, and hereditary persistence of fetal hemoglobin (HPFH). Different studies have assessed and concluded that HbF can interfere with HbA1c results.22 Detection of hemoglobin F, regardless of its level, is important because it can indicate not only HPFH but also the presence of thalassemia and delta beta thalassemia. Both conditions affect the normal hematology.
Methods that do not detect HbF and include its value in the formula for the calculation of HbA1c, are at risk of reporting underestimated A1c values for two reasons:
- The N-glycine terminal of hemoglobin F gamma chains doesn’t glycate at the same rate as the N-valine terminal of hemoglobin A beta chains. This difference may be explained because the N-glycine terminal of HbF is also subject to acetylation, which competes with glycation for the same site.22,31
- Methods that use the N-valine terminal epitope of the beta chain to bind antibodies and detect A1c are not able to bind and read the N-glycine terminal of the gamma chains.
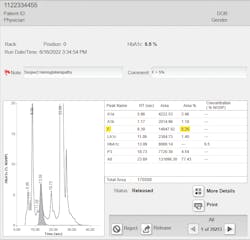
Furthermore, with these methods (IA, boronate affinity, enzymatic), laboratories are testing samples without the knowledge of the level of HbF (or variants) present in those samples.32 The advantage of cation-exchange high-performance liquid chromatography (CE-HPLC) as a widely used laboratory test is that it resolves the F peak from the A1c peak and doesn’t include HbF in the formula for the calculation of %A1c (see Figure 1).
Summary
When discussing the glycation of hemoglobin, the possible interferences that can influence this reaction, and the ability of the clinical laboratory to evaluate the %A1c value and diabetes control, it is important to consider the overall clinical condition of the subject and the possible natural occurring interactions of hemoglobinopathies. Reviewing the hematology presentation (RBC, RDW, MCV, MCH, hemolytic anemia) in combination with the A1c value is important in diabetes diagnostics.
Knowledge and awareness of thalassemia and hemoglobin variants affecting HbA1c measurements is essential, especially in areas with a high prevalence of hemoglobinopathy, to avoid the mismanagement of patients living with diabetes.2
References
- Rohlfing CL, Connolly SM, England JD, et al. The effect of elevated fetal hemoglobin on hemoglobin A1c results: five common hemoglobin A1c methods compared with the IFCC reference method. Am J Clin Pathol. 2008;129(5):811-814. doi:10.1309/YFVTUD0GHJF7D16H.
- Irene Shu et al. (2012) Comparison of hemoglobin A1c measurements of samples with elevated fetal hemoglobin by three commercial assays. Clin Chim Acta. Volume 413, Issues 19–20, Pages 1712-1713. doi: 10.1016/j.cca.2012.05.017.
- Little RR, Rohlfing CL, Hanson SE, et al. The effect of increased fetal hemoglobin on 7 common Hb A1c assay methods. Clin Chem. 2012;58(5):945-947. doi:10.1373/clinchem.2012.181933.
- Nitta T, Yamashiro Y, Hattori Y, Ezumi T, Nishioka M, Nakamura J. The interference by HbF on HbA1c (BM Test HbA1c) measurement in enzymatic method. Ann Clin Biochem. 2015;52(Pt 5):569-575. doi:10.1177/0004563214568872.
- Wambua S, Mwacharo J, Uyoga S, Macharia A, Williams TN. Co-inheritance of alpha+-thalassaemia and sickle trait results in specific effects on haematological parameters. Br J Haematol. 2006;133(2):206-209. doi:10.1111/j.1365-2141.2006.06006.x.
- el-Hazmi MA. Studies on sickle cell heterozygotes in Saudi Arabia--interaction with alpha-thalassaemia. Acta Haematol. 1986;75(2):100-104. doi:10.1159/000206095.
- Habara AH, Shaikho EM, Steinberg MH. Fetal hemoglobin in sickle cell anemia: The Arab-Indian haplotype and new therapeutic agents. Am J Hematol. 2017;92(11):1233-1242. doi:10.1002/ajh.24872.
- Fucharoen S, Weatherall DJ. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2012;2(8):a011734-a011734. doi:10.1101/cshperspect.a011734.
- A. A. Dani (2007) Double heterozygous for hemoglobin S and hemoglobin E — a case report from central India. Indian J Hematol Blood Transfus. 23(3-4): 119–121. doi: 10.1007/s12288-008-0012-0.
- Singha K, Fucharoen G, Fucharoen S. Five hemoglobin variants in a double heterozygote for α- and β-globin chain defects. Acta Haematol. 2014;131(2):71-75. doi:10.1159/000353123.
- Hoyer JD, Penz CS, Fairbanks VF, Hanson CA, Katzmann JA. Flow cytometric measurement of hemoglobin F in RBCs: diagnostic usefulness in the distinction of hereditary persistence of fetal hemoglobin (HPFH) and hemoglobin S-hPFH from other conditions with elevated levels of hemoglobin F: Diagnostic usefulness in the distinction of hereditary persistence of fetal hemoglobin (HPFH) and hemoglobin S-HPFH from other conditions with elevated levels of hemoglobin F. Am J Clin Pathol. 2002;117(6):857-863. doi:10.1309/A63X-HG9T-VYG2-X6TX.
- Efremov GD, Ibarra B, Gurgey A, Sukumaran PK, Altay C, Huisman TH. Gamma-chain heterogeneity of fetal hemoglobin in nonblack beta- and delta beta- thalassemia and HPFH heterozygotes and homozygotes. Am J Hematol. 1982;12(4):367-382. doi:10.1002/ajh.2830120408.
- A. Cao. Beta-Thalassemia Available at http://www.genetests.org. Accessed August 1, 2022.
- Mosca A, Paleari R, Leone D, Ivaldi G. The relevance of hemoglobin F measurement in the diagnosis of thalassemias and related hemoglobinopathies. Clin Biochem. 2009;42(18):1797-1801. doi:10.1016/j.clinbiochem.2009.06.023.
- Choudhary A, Giardina P, Antal Z, Vogiatzi M. Unreliable oral glucose tolerance test and haemoglobin A1C in beta thalassaemia major--a case for continuous glucose monitoring? Br J Haematol. 2013;162(1):132-135. doi:10.1111/bjh.12322.
- Old J. Hemoglobinopathies and Thalassemias. Emery and Rimoin’s Principles and Practice of Medical Genetics. 6th ed. Elsevier; 2013: 1-44.
- Sacks DB. Measurement of hemoglobin A(1c): a new twist on the path to harmony. Diabetes Care. 2012;35(12):2674-80. doi: 10.2337/dc12-1348.
- Cohen RM, Franco RS, Khera PK, Smith EP, Lindsell CJ, Ciraolo PJ, Palascak MB, Joiner CH. Red cell life span heterogeneity in hematologically normal people is sufficient to alter HbA1c. Blood. 2008 15;112(10):4284-91. doi: 10.1182/blood-2008-04-154112.
- Khera PK, Smith EP, Lindsell CJ, Rogge MC, Haggerty S, Wagner DA, Palascak MB, Mehta S, Hibbert JM, Joiner CH, Franco RS, Cohen RM. Use of an oral stable isotope label to confirm variation in red blood cell mean age that influences HbA1c interpretation. Am J Hematol. 2015;90(1):50-55. doi: 10.1002/ajh.23866.
- Guo W, Zhou Q, Jia Y, Xu J. Increased Levels of Glycated Hemoglobin A1c and Iron Deficiency Anemia: A Review. Med Sci Monit. 2019 7;25:8371-8378. doi: 10.12659/MSM.916719.
- Patel HH, Patel HR, Higgins JM. Modulation of red blood cell population dynamics is a fundamental homeostatic response to disease. Am J Hematol. 2015;90(5):422-8. doi: 10.1002/ajh.23982.
- Weykamp C. HbA1c: a review of analytical and clinical aspects. Ann Lab Med. 2013;33(6):393-400. doi: 10.3343/alm.2013.33.6.393.
- Kabytaev K, Connolly S, Rohlfing CL, Sacks DB, Stoyanov AV, Little RR. Higher degree of glycation of hemoglobin S compared to hemoglobin A measured by mass spectrometry: Potential impact on HbA1c testing. Clin Chim Acta. 2016 1;458:40-3. doi: 10.1016/j.cca.2016.04.027.
- Abraham EC, Cameron BF, Abraham A, Stallings M. Glycosylated hemoglobins in heterozygotes and homozygotes for hemoglobin C with or without diabetes. J Lab Clin Med. 1984;104(4):602-9.
- Abraham EC, Rao KR. Glycosylated hemoglobins in a diabetic patient with sickle cell anemia. Clin Physiol Biochem. 1987;5(6):343-9.
- Graffeo L, Vitrano A, Scondotto S, Dardanoni G, Pollina Addario WS, Giambona A, Sacco M, Di Maggio R, Renda D, Taormina F, Triveri A, Attanasio M, Gluud C, Maggio A. β-Thalassemia heterozygote state detrimentally affects health expectation. Eur J Intern Med. 2018 Aug;54:76-80. doi: 10.1016/j.ejim.2018.06.009.
- Guimarães JS, Cominal JG, Silva-Pinto AC, Olbina G, Ginzburg YZ, Nandi V, Westerman M, Rivella S, de Souza AM. Altered erythropoiesis and iron metabolism in carriers of thalassemia. Eur J Haematol. 2015 Jun;94(6):511-8. doi: 10.1111/ejh.12464.
- Galanello R. et al. (2005). Alpha-Thalassemia. Available at http://www.genetests.org. Accessed August 1, 2022.
- Schrier SL, Bunyaratvej A, Khuhapinant A, Fucharoen S, Aljurf M, Snyder LM, Keifer CR, Ma L, Mohandas N. The unusual pathobiology of hemoglobin constant spring red blood cells. Blood. 1997;89(5):1762-9.
- Fucharoen S, Viprakasit V. Hb H disease: clinical course and disease modifiers. Hematology Am Soc Hematol Educ Program. 2009:26-34. doi: 10.1182/asheducation-2009.1.26.
- Garlick RL, Shaeffer JR, Chapman PB, Kingston RE, Mazer JS, Bunn HF. Synthesis of acetylated human fetal hemoglobin. J Biol Chem. 1981 25;256(4):1727-31.
- Little RR, Rohlfing C, Sacks DB. The National Glycohemoglobin Standardization Program: Over 20 Years of Improving Hemoglobin A1c Measurement. Clin Chem. 2019 ;65(7):839-848. doi: 10.1373/clinchem.2018.296962.
About the Author

Marco Flamini MSc
Marco Flamini, MSc is a senior product manager for Bio-Rad Laboratories. He obtained his master’s degree in pharmacy from University La Sapienza in Italy. His focus is on hemoglobin testing and screening strategies. He was a member of the CLSI Document Development Committee on Newborn Screening for Hemoglobinopathies (NBS08).