The prospective use of cytokine markers for inflammatory bowel disease evaluation
For more information, visit the Continuing Education tab.
LEARNING OBJECTIVES
Upon completion of this article, the reader will be able to:
1. Describe and list the two Inflammatory Bowel Disease (IBD)forms.
2. Describe the relevance of Fecal Biomarkers for IBD diagnosisand assessment.
3. Describe the relevance of Autoantibody markers for IBDphenotyping.
4. Describe the relevance of Cytokine Signatures for IBDdiagnosis and evaluation.
Inflammatory bowel disease (IBD) is a group of chronic inflammatory diseases that are differentiated by the location and type of inflammation. Ulcerative colitis (UC) is characterized by chronic, continuous, ulcerative inflammation limited to the inner linings of the colon and predominantly the rectum. Crohn’s disease (CD) is described as chronic, patchy inflammation anywhere in the gastrointestinal tract. Patients typically experience mild to severe gastrointestinal-related symptoms in irregular cycles of active illness and remission.1
IBD pathophysiology
It is hypothesized that IBD is the result of increased permeability of the gut epithelial barrier. Normally, a thick layer of mucus and selective tight junctions between epithelial cells regulate paracellular commensal antigen trafficking from the intestinal lumen to the gut immune system. However, an impaired barrier will introduce commensal bacterial antigens to intestinal immune cells, inducing an immune response of pro-inflammatory and lymphocyte-activating cytokines.2 The inflammatory markers increase intestinal epithelial permeability and expose more commensal antigens to the immune system, secreting subsequential inflammatory markers.3 IBD is associated with increased intestinal permeability, hyper belligerent immune response, and change in the gut microbiome.
Currently, the most prominent theory held is dysbiosis: an overall increase of gut bacteria that induce inflammation and a reduction of gut bacteria that mitigate inflammation.4
Ulcerative colitis
The intestinal composition of UC patients is imbalanced in population, stability, and diversity. Popular short chain fatty acid (SCFA) producers, Faecalibacterium prausnitzii (F. prausnitzii), Roseburia spp., Clostridia spp., and Akkermansia spp. are significantly depleted in UC patients.5, 6 Depleted energy-rich SCFAs are associated with intestinal epithelial nutrient deficiency, severe inflammatory response, and intestinal environments where pathogenic facultative anaerobes can thrive.
Crohn’s disease
CD includes a reduction in protective, obligate anaerobes. From the Firmicute phylum, CD patients have reduced F. prausnitzii, Roseburia inulinivorans, Ruminococcus torques, and Clostridium lavalense.4, 7 The loss of butyrate-producing F. prausnitzii is related to recurrence of CD; transplantation of F. prausnitzii appears to reduce inflammation.8
There are a negligible number of differences in microbiota in UC and CD. However, UC patients consistently have higher heterogeneity in microbial signatures compared to CD,9 and as a result, it makes it difficult to identify UC purely based on the microbial composition. Disruptions in enteric microbiota give rise to subtle alterations in the immune response, so clinicians can measure the byproducts of these modified responses to assist them in the diagnosis of IBD.
IBD laboratory testing
Endoscopic procedures remain the gold standard for diagnosing IBD. This invasive procedure examines the interior cavities of the small intestine and colon and grades the severity of intestinal inflammation on a semiquantitative scale. Alternatively, computed tomography and magnetic resonance imaging scans are imaging techniques for diagnosing, grading, and accurately discriminating between the IBD subtypes.10 Histological examination is another substitute, but it requires a biopsy and lacks a singular definitive histological feature that differentiates between subtypes.11 Nonetheless, these imaging procedures are expensive, time-consuming, and resource-intensive. Endoscopy is usually avoided by the pediatric population for its discomfort and in severe IBD cases where analysis can risk ulcer perforation.
Currently, there is no definitive blood biomarker capable of identifying IBD, discriminating between its subtypes, grading its severity, and determining its likelihood of remission comparatively to an endoscopic procedure. Some nonspecific inflammatory markers can assist in diagnosing IBD. Elevated C-Reactive Protein (CRP), erythrocyte sedimentation rate (ESR), white blood cell count, platelets, and albumin levels allude to inflammation.11 CRP is a nonspecific marker for inflammation that is significantly increased in CD and moderately increased in UC. CRP is better tailored to CD, for it can distinguish between mild to moderate CD and identify quiescent CD.12 Elevated circulating fibrocytes are positively correlated with CD severity,13 but this increase is nonspecific to gastrointestinal tract wounds.
Fecal markers
Stool is an accessible, noninvasive patient specimen that is reflective of gastrointestinal tract activity. The presence of certain metabolites may indicate infection or inflammatory activity. Stool is stable at room temperature, resistant to multiple freezing and thawing cycles, and easily processable. Some stool metabolites have approved quantitative point of care test kits.14
Fecal calprotectin
Calprotectin is released by neutrophils at an inflamed site, so fecal measurement of calprotectin is an indirect measurement of neutrophil infiltration of the gastrointestinal tract.15 CD stool specimens have four times the mean fecal calprotectin (FC) compared to the healthy stool; high FC is associated with reduced microbial diversity in severely inflamed patients.9 FC can detect lower inflammatory activity better than CRP and can distinguish between active versus quiescent IBD and IBD versus irritable bowel syndrome (IBS).16 Compared to all other noninvasive procedures, FC dominates with a 64% to 95% sensitivity and a 79% to 93% specificity.17 Furthermore, unlike CRP, FC can grade the severity of most IBD subtypes.
In determining postoperative recurrence, an elevated FC biomarker greater than 120 µg/g is a better predictor of endoscopic scores than CRP and ESR.18 Within one year, FC can predict a relapse in post-surgical CD patients with an 83% sensitivity and 93% specificity.14 FC is capable of discriminating between major CD and UC subtypes. For example, ileocolonic and colonic CD have significantly higher FC than terminal CD.16 FC measurements can distinguish between pouchitis, cuffitis, and CD with a 92% specificity.14
However, FC is incapable of identifying all CD subtypes, like small bowel CD.17 FC lacks absolute specificity, for it can be falsely elevated if a patient is taking anti-inflammatory medications or has an intestinal infection. Furthermore, there is increased variability in consecutive same patient samples, for day-to-day changes in diet and physical activity can shift FC indices.12 There is not enough data to support FC as a comparative substitute to endoscopy, but it remains to be a useful, noninvasive diagnostic test to assess intestinal inflammation.
Fecal lactoferrin
Changes in fecal lactoferrin (FL) levels are proportional with leukocyte margination and diapedesis into the gut mucosa and exhibit a close correlation with inflammatory endoscopic and histological activity.19, 20 Comparable to histologic analysis, FL was reported to increase within 2 months of pouchitis development.
CD patients who maintained elevated FL levels post-surgical intervention experienced relapse within 4 years of operation, suggesting that subclinical inflammation persisted. In post-operative CD patients who relapsed within a year of intervention, FL concentrations were consistently doubled in all cases in comparison to their base levels.14
It has been shown that FL is capable of distinguishing, with a 95% specificity, between a healthy and an IBD patient. However, FL is a weak discriminator against other gastrointestinal inflammatory diseases, differentiating between IBD and IBS with only a 61% specificity.19 Another limitation is that FL is incapable of detecting quiescent IBD.21 A definitive FL cutoff value is inconsistent among current literature.22 Researchers suggest pairing FL with other confirmatory tests, like a colonoscopy.23 Some studies argue that the diagnostic precision of FL is inferior to FC. However, there are more studies on FC as a biomarker for IBD than FL, so this argument remains inconclusive.
Fecal microbiota
IBD is more associated with the loss of beneficial microorganisms, like Faecalibacterium spp., Methanobrevibacter spp., and Oscillospira spp, rather than the gain of pathogenic bacteria, like Desulfovibrio spp.23 Elevations in the Eubacteriaceae, Bifidobacteriaceae, and Lachnospiraceae families are associated with signs of remission. If either of these bacterial strains are depleted, then T-regulatory (Treg) cells homeostasis and regulated T-cells differentiation are compromised.24 Potent butyrate metabolizers, like Roseburia intestinalis, and tryptophan metabolizers, like Clostridium sporogenes and beneficial E. coli, are reduced in stool, priming the intestine for inflammation.25
Stool specimens are not completely reflective of microbial perturbance. Deep bacteria and bacteria that strongly adhere to the epithelial lining are excluded in stool specimens. Furthermore, the distinctive fecal microbial signatures of all IBD subtypes need to be mapped in the future.26
Autoantibodies
The presence of autoantibodies suggests a loss of tolerance to commensal bacteria. Thus, the reduction of intestinal bacteria in IBD affects the proper development of gut immune tolerance.5
Antineutrophil Cytoplasmic Antibody
Antineutrophil cytoplasmic antibody (ANCA) is categorized based on two staining patterns: cytoplasmic ANCA and peripheral ANCA (pANCA).27
26.19% of CD and 66.05% of UC individuals are ANCA positive which show that not all IBD cases will produce ANCA.28 A positive pANCA test has an 86% specificity to UC patients and 66% specificity to CD patients, suggesting that pANCA is a poor IBD screening test but a strong UC discriminator.29
Although ANCA is a potent UC discriminator, no evidence suggests that ANCA can pinpoint the location of inflammatory involvement nor can pANCA predict postoperative relapse, proving this marker to be inferior to endoscopy.29, 30
Anti-Saccharomyces cerevisiae antibody
Anti-Saccharomyces cerevisiae Antibody (ASCA) was initially discovered to target the cell wall of Saccharomyces cerevisiae, but it is now observed in some instances of IBD. S. cerevisiae yeast can be normally found in healthy intestines and is reported to have protective antimicrobial properties.25
When evaluated on its ability to discriminate CD from other IBD subtypes, ASCA demonstrated a 16% sensitivity and 97% specificity, suggesting that ASCA is a poor IBD screening test but a strong CD discriminator.29 Unlike ANCA, increasing ASCA titers predicted relapse and fistulae development in CD patients.31
Similar to ANCA, there is currently no evidence that suggests ASCA can pinpoint the location of inflammation.30 Moreover, ASCA positivity is only detectable in 20% of CD cases.32

Cytokine markers
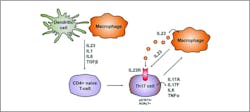
IBD is a poorly controlled immune response caused by gut perturbance. The significant reduction in number and type of microorganisms limit proper immune tolerance development. As a result, effector T cells are overreactive and Treg cells are ineffective in maintaining gut homeostasis.33 Imbalance of interleukins (IL), interferons (IFN), tumor necrosis factor (TNF), and transforming growth factor (TGF) cytokines account for crudely regulated immune system. Chemokines also play a major role in IBD; they recruit more immune cells to destroy the epithelial lining, leading to the loss of gut integrity, function, and invasion of the lamina propria.34
UC and CD have distinctive cytokine profiles; elevated T-helper 2 (Th2) cell related cytokines, such as IL-13, IL-5, and IL-9, and elevated T-helper 1 (Th1) cell related cytokines, such as IL-12, IL-23, and IFN-y, are indicative of UC and CD, respectively.34 The subtypes’ identifiable cytokine profiles are antagonistic to one another. Th1 cell-mediated CD can downregulate Th2 cell-mediated UC with IFN-y, and Th2 cell-mediated UC can downregulate Th1 cell-mediated CD with IL-4 and IL-13.33 Both IBD subtypes have a markedly elevated immune mediators, including TNF-α, IFN-γ and IL-6,34 which correlate to the development of severe mucosal inflammation. In UC and CD, IL-4 and lamina propria-healing TGF-β is reduced; a reduction of TGF-β is linked with the development of several autoimmune diseases.35 Cytokine expression profiles can help identify disease onset, exacerbation, and phenotype.
Cytokines associated with ulcerative colitis
UC is described as an atypical Th2 cell-mediated chronic autoimmune response. Pro-inflammatory IL-33 is consistently increased in UC.36 IL-33 is elevated because it promotes Th2 cell activity and eosinophil infiltration while suppressing Th1 and T-helper 17 (Th17) cell activity.34 IL-35, an inducer for T-cell differentiation to Treg and B-cell differentiation to regulatory B cells, is reduced.
Interestingly, anti-inflammatory IL-10 is elevated in UC which inhibits macrophage and lymphocyte activity and downregulates pro-inflammatory cytokines. Although appearing counterintuitive, it is hypothesized that this increase is the immune system’s failed attempt to counter the pro-inflammatory cytokine dysregulation and return to intestinal homeostasis.37
Cytokines associated with crohn’s disease
An atypical Th1 cell immune response promotes intracellular killing through activated macrophages and cytotoxic lymphocytes. IL-12 indirectly induce IFN-γ, favoring Th1 cell upregulation and overexpression. Higher levels of IL-18 are seen in CD patients with chronic lesions compared to healthy controls. Elevated IL-18 can help proliferate Th1 cells and synergize with IL-12 to promote IFN-γ production.35
Th17 cells secrete the IL-17 cytokine family which have both pro-inflammatory and tissue-protective attributes.34 As CD progresses from early mucosal inflammation to established lesions, pro-inflammatory IL-17A is markedly increased, suggesting Th17 cell’s active role in the pathogenesis and disease progression of CD.38
Dysbiosis in the gut can also play a role in the expression of Th1 cells and the downregulation of Treg cells. A depleted Clostridium spp. and Bacteroides spp. and abundant segmented filamentous bacteria (SFB) create a microbial environment that favors the stimulation of pro-inflammatory cells. Reduced Clostridium clusters IV, XIVa and XVII, B. fragilis, and F. prausnitzii blunt bacterial antigenic signals that trigger Treg cells expansion.39 On the opposite end, SFB can produce bacteria-derived adenosine triphosphate which can activate colonic lamina propria cells and stimulate pro-inflammatory cytokine release from Th1 and Th17 cells.33
Cytokines signatures as a diagnostic marker
Upon microbial perturbance and gut immune system exposure to disrupted commensal bacteria, immunoregulatory Treg cells can translate into pro-inflammatory Th1 or Th17 effector cells and initiate mucosal inflammation.40 After cytokine feedback, Th17 cells may also revert back to Treg cells through a property called T-cell plasticity.38
Complex cytokine networks interact with epithelial and immune cells to maintain intestinal homeostasis and immunotolerance. For example, IL-22 and IL-6 coupled with IL-17 can activate signal transducer and activator of transcription 3, prompting colonic epithelial cell survival and stimulating their antimicrobial properties.41 Additionally, gut bacteria and diet metabolites induce IL-10 and TGF-β1 production, cytokines that play a key role in developing tolerance for mononuclear phagocytic cells. Clinicians can pinpoint the beginning of colitis manifestation by identifying mononuclear phagocytic cell secretion of IL-1β, IL-18, and TNF-α. These cells also drive Treg cell promotion.41 If any part of this pathway is disrupted, then it may lead to autoimmunity against intestinal epithelial cells.42
Th17 cells are initially developed in the presence of low TGF-β and IL-1β levels followed by the expansion of the population through IL-21 activation. Finally, IL-23 stabilizes and maintains the developed Th17 cell population.43 IL-23, the main Th17 cells initiating cytokine, drives intestinal inflammation and chronic colitis by inhibiting Treg cell expansion and inducing naïve CD4+ T-cells differentiation into Th17 cells.41 High IL-23 serum concentrations are associated with worsened disease severity and longer duration of UC,44 and deficiencies in the IL-23 receptor gene is associated with IBD development.40
The disease stages of CD can be predicted through the Th1 to Th17 cell ratio. Early CD is dominated by Th1 cell derived cytokines like IFN-γ, but developed CD is identified as a mix of both Th1 cell and Th17 cell derived cytokines, exemplifying T-cell plasticity. Higher levels of IL-17A and IL-22 are identifiable in later stages of CD.45 Pro-inflammatory IL-21 is also upregulated at this stage. IL-21 expands Th1, inhibits Treg cells differentiation, and makes CD4+ T-cells resistant to Treg cells mediated immune suppression.46 Measuring the Treg to Th17 cells ratio can potentially assist in grading the extent of colonic inflammation.45
The IL-17/IL-23 cytokine axis is linked to several chronic inflammatory diseases. In theory, IL-23 can indirectly stimulate IL-17 serum levels and subsequently initiate and prolong intestinal inflammation. IL-23 serum levels are a reliable predictor for intestinal manifestations, including enteral fistulae and abscess development, and are associated with UC disease severity and duration.44 The gene associated for coding the IL-23 receptor is overexpressed in CD.38, 47
Similar to IL-23, IL-17 is most prominently found in the submucosa and muscularis propria of CD and UC patients. IL-17 and IL-23 serum concentrations can potentially grade disease severity. IL-23 is significantly elevated in severe CD and UC while IL-17 is only significantly elevated in severe UC. In severe CD, IL-23 had a 100% sensitivity and 100% specificity, respectively, and IL-17 had a 53.33% sensitivity and 87.50% specificity, respectively. In the severe UC cohort, IL-23 had a 94.12% sensitivity and 100% specificity, respectively. IL-17 had 94.17% sensitivity and 100% specificity, respectively.44 The box-and-whiskers plot in figure 2 displays elevated IL-17 in severe UC and elevated IL-23 in severe UC and CD. The discriminatory potential of IL-23 towards IBD subtype severity is superior to FC, a frequently used IBD-monitoring assay, with a p-value of < 0.001.44 Elucidation of the IL-17/IL-23 axis may change the understanding of IBD pathogenesis and can become a potential adjunct IBD diagnostic marker.
Conclusion
The diagnostic potential of FC and FL triumph over nonspecific CRP and ESR assays. Although favorable and reflective of enteric perturbance, fecal microbiota signatures require further investigation. ANCA and ASCA are potent UC and CD discriminators, respectively, but they lack the sensitivity to be practical. UC and CD are predominantly characterized by Th2 cells and Th1 cells derived cytokines, respectively. Treg cells inhibit inflammatory T-cell differentiation, so Treg to Th17 cell ratio yields a promising future for grading the extent of inflammation. Cytokine signatures, like the IL-17/IL-23 axis, may play a prolific role as a potential adjunct diagnostic tool for IBD. Early stages of CD are identifiable through the prevalence of Th1 cells derived cytokines, and developed stages of CD are identifiable through the prevalence of both Th1 cells and Th17 cells derived cytokines. IL-17 and IL-23 prove to be potent blood biomarkers for identifying both severe UC and CD. Further investigation should explore the dynamic progression and balance of cytokine and chemokine levels throughout the IBD course.
References
- Inflammatory bowel disease (IBD). Mayo Clinic. Published November 7, 2020. Accessed June 3, 2022. http://www.mayoclinic.org/diseases-conditions/inflammatory-bowel-disease/symptoms-causes/syc-20353315.
- Becker C, Neurath MF, Wirtz S. The intestinal microbiota in inflammatory bowel disease. ILAR J. 2015;56(2):192-204. doi:10.1093/ilar/ilv030.
- Fasano A. All disease begins in the (leaky) gut: role of zonulin-mediated gut permeability in the pathogenesis of some chronic inflammatory diseases. F1000Res. 2020;9:69. doi:10.12688/f1000research.20510.1.
- Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. 2018;11(1):1-10. doi:10.1007/s12328-017-0813-5.
- Vangoitsenhoven R, Cresci GAM. Role of microbiome and antibiotics in autoimmune diseases. Nutr Clin Pract. 2020;35(3):406-416. doi:10.1002/ncp.10489.
- Du L, Ha C. Epidemiology and pathogenesis of ulcerative colitis. Gastroenterol Clin North Am. 2020;49(4):643-654. doi:10.1016/j.gtc.2020.07.
- Major G, Spiller R. Irritable bowel syndrome, inflammatory bowel disease and the microbiome. Curr Opin Endocrinol Diabetes Obes. 2014;21(1):15-21. doi:10.1097/MED.0000000000000032.
- Durack J, Lynch SV. The gut microbiome: Relationships with disease and opportunities for therapy. J Exp Med. 2019;216(1):20-40. doi:10.1084/jem.20180448.
- Franzosa EA, Sirota-Madi A, Avila-Pacheco J, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4(2):293-305. doi:10.1038/s41564-018-0306-4.
- Kilcoyne A, Kaplan JL, Gee MS. Inflammatory bowel disease imaging: current practice and future directions. World J Gastroenterol. 2016;22(3):917-932. doi:10.3748/wjg.v22.i3.917.
- Shokrani M. Inflammatory bowel disease: diagnosis and research trends: the clinical lab is playing an increasingly important role. MLO Med Lab Obs. 2012;44(8):8, 10, 12; quiz 14.
- Stragier E, Van Assche G. The use of fecal calprotectin and lactoferrin in patients with IBD. Review. Acta Gastroenterol Belg. 2013;76(3):322-328.
- Kiernan MG, Coffey JC, Sahebally SM, et al. Systemic molecular mediators of inflammation differentiate between Crohn’s disease and ulcerative colitis, implicating threshold levels of IL-10 and relative ratios of pro-inflammatory cytokines in therapy. J Crohns Colitis. 2020;14(1):118-129. doi:10.1093/ecco-jcc/jjz117.
- Caccaro R, Angriman I, D’Incà R. Relevance of fecal calprotectin and lactoferrin in the post-operative management of inflammatory bowel diseases. World J Gastrointest Surg. 2016;8(3):193-201. doi:10.4240/wjgs.v8.i3.193.
- Hejl J, Theede K, Møllgren B, et al. Point of care testing of fecal calprotectin as a substitute for routine laboratory analysis. Pract Lab Med. 2018;10:10-14. doi:10.1016/j.plabm.2017.11.002.
- Lee YW, Lee KM, Lee JM, et al. The usefulness of fecal calprotectin in assessing inflammatory bowel disease activity. Korean J Intern Med. 2019;34(1):72-80. doi:10.3904/kjim.2016.324.
- Vernia F, Di Ruscio M, Stefanelli G, Viscido A, Frieri G, Latella G. Is fecal calprotectin an accurate marker in the management of Crohn’s disease? J Gastroenterol Hepatol. 2020;35(3):390-400. doi:10.1111/jgh.14950.
- Quest Diagnostics: Calprotectin, Stool. Questdiagnostics.com. Accessed June 3, 2022. http://testdirectory.questdiagnostics.com/test/test-detail/16796/calprotectin-stool?p=r&cc=MASTER.
- Wang Y, Pei F, Wang X, Sun Z, Hu C, Dou H. Diagnostic accuracy of fecal lactoferrin for inflammatory bowel disease: a meta-analysis. Int J Clin Exp Pathol. 2015;8(10):12319-12332.
- Rubio MG, Amo-Mensah K, Gray JM, et al. Fecal lactoferrin accurately reflects mucosal inflammation in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2019;10(5):54-63. doi:10.4291/wjgp.v10.i5.54.
- Zhou XL, Xu W, Tang XX, et al. Fecal lactoferrin in discriminating inflammatory bowel disease from irritable bowel syndrome: a diagnostic meta-analysis. BMC Gastroenterol. 2014;14(1):121. doi:10.1186/1471-230X-14-121.
- Liu F, Lee SA, Riordan SM, Zhang L, Zhu L. Global studies of using fecal biomarkers in predicting relapse in inflammatory bowel disease. Front Med (Lausanne). 2020;7:580803. doi:10.3389/fmed.2020.580803.
- Lopez RN, Leach ST, Lemberg DA, Duvoisin G, Gearry RB, Day AS. Fecal biomarkers in inflammatory bowel disease: Faecal biomarkers. J Gastroenterol Hepatol. 2017;32(3):577-582. doi:10.1111/jgh.13611.
- Papa E, Docktor M, Smillie C, et al. Non-invasive mapping of the gastrointestinal microbiota identifies children with inflammatory bowel disease. PLoS One. 2012;7(6):e39242. doi:10.1371/journal.pone.0039242.
- Liu S, Zhao W, Lan P, Mou X. The microbiome in inflammatory bowel diseases: from pathogenesis to therapy. Protein Cell. 2021;12(5):331-345. doi:10.1007/s13238-020-00745-3.
- Pascal V, Pozuelo M, Borruel N, et al. A microbial signature for Crohn’s disease. Gut. 2017;66(5):813-822. doi:10.1136/gutjnl-2016-313235.
- Mitsuyama K, Niwa M, Takedatsu H, et al. Antibody markers in the diagnosis of inflammatory bowel disease. World J Gastroenterol. 2016;22(3):1304-1310. doi:10.3748/wjg.v22.i3.1304.
- Li D, Silverberg MS, Haritunians T, et al. TNFRSF1B is associated with ANCA in IBD. Inflamm Bowel Dis. 2016;22(6):1346-1352. doi:10.1097/MIB.0000000000000771.
- Mokhtarifar A, Ganji A, Sadrneshin M, et al. Diagnostic value of ASCA and atypical p-ANCA in differential diagnosis of inflammatory bowel disease. Middle East J Dig Dis. 2013;5(2):93-97.
- Pang Y, Ruan H, Wu D, Lang Y, Sun K, Xu C. Assessment of clinical activity and severity using serum ANCA and ASCA antibodies in patients with ulcerative colitis. Allergy Asthma Clin Immunol. 2020;16(1):37. doi:10.1186/s13223-020-00433-1.
- Coukos JA, Howard LA, Weinberg JM, Becker JM, Stucchi AF, Farraye FA. ASCA IgG and CBir antibodies are associated with the development of Crohn’s disease and fistulae following ileal pouch-anal anastomosis. Dig Dis Sci. 2012;57(6):1544-1553. doi:10.1007/s10620-012-2050-6.
- Plevy S, Silverberg MS, Lockton S, et al. Combined serological, genetic, and inflammatory markers differentiate non-IBD, Crohn’s disease, and ulcerative colitis patients. Inflamm Bowel Dis. 2013;19(6):1139-1148. doi:10.1097/MIB.0b013e318280b19e.
- Silva FAR, Rodrigues BL, Ayrizono M de LS, Leal RF. The immunological basis of inflammatory bowel disease. Gastroenterol Res Pract. 2016;2016:2097274. doi:10.1155/2016/2097274.
- Nemeth ZH, Bogdanovski DA, Barratt-Stopper P, Paglinco SR, Antonioli L, Rolandelli RH. Crohn’s disease and ulcerative colitis show unique cytokine profiles. Cureus. 2017;9(4):e1177. doi:10.7759/cureus.1177.
- Moldoveanu AC, Diculescu M, Braticevici CF. Cytokines in inflammatory bowel disease. Rom J Intern Med. 2015;53(2):118-127. doi:10.1515/rjim-2015-0016.
- Guan Q, Zhang J. Recent advances: the imbalance of cytokines in the pathogenesis of inflammatory bowel disease. Mediators Inflamm. 2017;2017:1-8. doi:10.1155/2017/4810258.
- Rodríguez-Perlvárez ML, García-Sánchez V, Villar-Pastor CM, et al. Role of serum cytokine profile in ulcerative colitis assessment. Inflamm Bowel Dis. 2012;18(10):1864-1871. doi:10.1002/ibd.22865.
- Schmitt H, Neurath MF, Atreya R. Role of the IL23/IL17 pathway in Crohn’s disease. Front Immunol. 2021;12:622934. doi:10.3389/fimmu.2021.622934.
- Khan I, Ullah N, Zha L, et al. Alteration of gut Microbiota in inflammatory bowel disease (IBD): Cause or consequence? IBD treatment targeting the gut microbiome. Pathogens. 2019;8(3):126. doi:10.3390/pathogens8030126.
- Ueno A, Ghosh A, Hung D, Li J, Jijon H. Th17 plasticity and its changes associated with inflammatory bowel disease. World J Gastroenterol. 2015;21(43):12283-12295. doi:10.3748/wjg.v21.i43.12283.
- Friedrich M, Pohin M, Powrie F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity. 2019;50(4):992-1006. doi:10.1016/j.immuni.2019.03.017.
- Sylvester FA, Draghi A, Menoret A, Fernandez ML, Wang Z, Vella AT. Distinctive colonic mucosal cytokine signature in new-onset, untreated pediatric Crohn disease. J Pediatr Gastroenterol Nutr. 2014;59(5):553-561. doi:10.1097/MPG.0000000000000480.
- Cătană CS, Berindan Neagoe I, Cozma V, Magdaş C, Tăbăran F, Dumitraşcu DL. Contribution of the IL-17/IL-23 axis to the pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2015;21(19):5823-5830. doi:10.3748/wjg.v21.i19.5823.
- Lucaciu LA, Ilieș M, Vesa Ștefan C, et al. Serum interleukin (IL)-23 and IL-17 profile in inflammatory bowel disease (IBD) patients could differentiate between severe and non-severe disease. J Pers Med. 2021;11(11):1130. doi:10.3390/jpm11111130.
- Zorzi F, Monteleone I, Sarra M, et al. Distinct profiles of effector cytokines mark the different phases of Crohn’s disease. PLoS One. 2013;8(1):e54562. doi:10.1371/journal.pone.0054562.
- Monteleone I, Pallone F, Monteleone G. Th17-related cytokines: new players in the control of chronic intestinal inflammation. BMC Med. 2011;9(1):122. doi:10.1186/1741-7015-9-122.
- Norouzinia M, Chaleshi V, Alizadeh AHM, Zali MR. Biomarkers in Inflammatory Bowel Diseases: Insights into Diagnosis, Prognosis, and Treatment. Gastroenterology and Hepatology from Bed to Bench.; 2017.
Dioco Reyes, MLS is a graduate of Medical Laboratory Sciences Program at Northern Illinois University and is a medical laboratory scientist at Advocate Good Samaritan Hospital in Downers Grove, IL.
Masih Shokrani, PhD MT (ASCP) is a professor in the Medical Laboratory Sciences Program at Northern Illinois University. He teaches medical immunology, medical diagnostic biochemistry and research.
About the Author

Dioco Reyes, MLS (ASCP)
is a graduate of Medical Laboratory Sciences Program at Northern Illinois University and is a medical laboratory scientist at Advocate Good Samaritan Hospital in Downers Grove, IL.