Novel and classical insights into Lp(a) concentration and the effects on various cardiovascular conditions
Despite advances in understanding and technology, cardiovascular diseases (CVDs) remain a major source of mortality across the world. The World Health Organization (WHO) estimates that 17.9 million people died due to CVDs in 2019, accounting for around 32% of deaths that year.1 First described in 1963, lipoprotein(a) [Lp(a)] is a macromolecular lipoprotein complex2 that is thought to display proatherogenic, proinflammatory,3 and prothrombotic4 potential and is considered an independent causal risk factor for various types of CVD.5 These properties provide several mechanisms in which elevated Lp(a) levels may contribute to CVD, however, the true nature of the Lp(a) relationship to CVD remains largely enigmatic.
Lp(a) concentrations in plasma are principally regulated by variation in LPA gene and levels remain relatively stable throughout one’s lifetime with lifestyle factors having little effect on their concentration.6 Due to the highly heritable nature of Lp(a) concentration, those with a family history of familial hypercholesterolaemia (FH), elevated Lp(a) levels, or atherosclerotic cardiovascular disease (ASCVD) should be screened, their plasma Lp(a) concentration determined, and their risk of CVD established.
In the last ten years, there have been many advances in the understanding of this ambiguous lipoprotein, which support the causal association with CVD, clarify the established evidence, and introduce novel mechanisms of action in relation to Lp(a), shedding light on its obscure pathophysiology. However, there are still diagnostic complications associated with Lp(a) measurement as there is little standardization in methods of determination.5
Physiology and genetics
Synthesised mainly in the liver, Lp(a), like LDL, is composed of a lipid centre made of cholesteryl esters and triacylglycerols, surrounded by a shell of phospholipids, free cholesterol, and an apoB-100 molecule. The major difference between other LDL molecules and Lp(a) is the presence of a polymorphic glycoprotein, apo(a), bound to apoB-100 by a single disulphide bond.5 It is this apo(a) molecule that contributes to Lp(a)s pathophysiology.
Apo(a) is thought to have evolved from the plasminogen gene (PLG) around 40 million years ago and shares 78–100% sequence homology within the untranslated and coding regions of the fibrinolytic enzyme.2 Like plasminogen, apo(a) contains unique domains named kringles.5 While plasminogen contains five different kringle structures (KI to KV), apo(a) has lost KI to KIII and instead contains several forms of KIV, namely, one copy of KIV-1, one copy of KIV3-10, 1–40 copies of KIV-2, one copy of KV, and an inactive protein domain at the carboxyl terminus of the molecule.7 These hydrophilic subunits are highly polymorphic due to the variation in KIV-2 repeats. Individuals may possess two different isoforms of apo(a) — one of which will have been passed down from each parent, which are expressed codominantly.2 These isoforms are dependent on the number of KIV-2 repeats they contain.2 Isoforms with less KIV-2 repeats produce smaller apo(a) isoforms, which are found at a higher concentration compared with larger isoforms8 due to the increased rate at which the smaller molecules can be synthesised.5 The polymorphisms in KIV-2 repeats account for up to 70% of the variation seen in concentration between individuals, with the remainder being attributed to differences in protein folding, transport, and single nucleotide polymorphisms (SNPs).5 SNPs are central in the heterogeneity of apo(a), effecting RNA splicing, nonsense mutations, and 5’ untranslated region of the LPA gene resulting in shorter gene translation.5,8
Pathophysiology
Lp(a) is thought to contribute to the risk of CVD through multiple mechanisms. Firstly, Lp(a) molecules display all the same atherosclerotic risk as LDL-C molecules due to their similar fundamental composition; for example, their propensity for oxidization upon entering the vessel wall and promotion of atherosclerosis through inflammatory and immunogenic mechanisms.9 (See Figure 1.) However, Lp(a) displays more proatherogenic potential due to the presence of the apo(a) molecule. The structure of apo(a) results in decreased fibrinolysis. Due to its structural similarities, apo(a) competes with plasminogen for binding sites, competitively inhibiting plasminogen, ultimately resulting in reduced fibrinolysis.9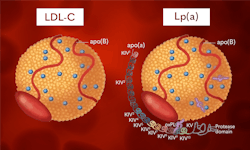
Lp(a) is thought to be a preferential carrier of oxidized phospholipids2 (OxPLs), which covalently bind to apo(a), increase expression of inflammatory proteins, and stimulate the secretion of IL-8 and monocyte chemoattractant protein-1, enhancing its ability to cross the vessel wall9 Some claims require further investigation, however, studies have been carried out that show inhibition of plasminogen activation in the presence of Lp(a).10 It is this indirect mechanism that Lp(a) is thought to conduct its prothrombotic activity.8,9
Clinical evidence
Many studies have been carried out to determine the association of Lp(a) concentration and CVD risk. Studies such as the Copenhagen General Population Study, the Copenhagen City Heart Study, Dallas Heart Study, and Ischemic Heart Disease Studies provide strong evidence for Lp(a) as a causal risk factor for CVD. Data analysis of the Copenhagen General Population Study reveal that 20% of subjects displayed Lp(a) concentrations of more than 42mg/dl, or around 105nmol/L,11 which is considered to result in increased risk of atherosclerotic disease.5 It is important to note, there is no accepted conversion factor for converting Lp(a) concentration from mg/dl to nmol/L due to the variability of apo(a) kringles. The unitage will depend on the assay method used.5 Another study in a healthcare organization in Israel showed that myocardial infarction (MI) and coronary artery disease were 2.5 times more common in those with high levels of Lp(a) than in the age and sex matched control group.3 This study,3 along with others,5,6,12 describe a linear relationship between Lp(a) concentration and CVD risk, showing at least a three-fold increase in ASCVD and MI events in adults with Lp(a) concentrations in the top 1% when compared with those with concentrations in the bottom 20%.3
The major variation in Lp(a) concentration seen throughout the population is further evident between ethnicities and sexes. On average, Caucasian subjects display the lowest Lp(a) concentrations, with Black subjects displaying the highest concentrations.5 However, the large number of functional variants are consistent across ethnicities suggesting that it is the KIV-2 repeats and SNPs that are the major factors contributing to Lp(a) concentration regardless of ethnicity. Lp(a) concentrations are higher in women than men8 with levels increasing post-menopause thought to be caused by a decrease in oestrogen.3
Testing and screening
The European Atherosclerosis Society (EAS) recommend that all adults are tested at least once in their lifetime to identify individuals who have high levels of Lp(a) and therefore high CVD risk. Screening is also recommended in children who have a family history of ischaemic stroke, premature ASCVD, or high Lp(a) levels in the absence of other identifiable risk factors.8 Testing has been associated with reduced mortality rates. This is thought to be because of increased and intensified therapy for those who are identified as high risk due to high plasma Lp(a) concentration.6
There are various assays available for the determination of Lp(a) concentration, which vary in accuracy and precision. Many of these assays utilize polyclonal antibodies which recognize different antigenic determinants.8 Due to the variability in apo(a) structure and KIV repeats, these assays often overestimate the concentration of large isoforms and underestimate concentration of small isoforms when determining the true Lp(a) levels.9 This variation can be partially nullified by using a calibrator series and by selecting a method that is traceable to WHO and International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) reference materials. This allows laboratories to confidently identify individuals considered high risk but may still prove problematic when patients’ results report closer to the assay thresholds.
One study13 compared five commercially available Lp(a) assays on an automated clinical chemistry analyzer. The assays tested were manufactured by Diazyme, Kamiya, MedTest, Roche, and Randox. The authors show that all the assays tested met the manufacturers’ claims for sensitivity, linearity, and precision. However, significant bias was observed in 4 out of 5 assays. The only assay that did not display significant bias was the Randox Lp(a) Assay, which is traceable to WHO/IFCC reference material. This report highlights the importance of measuring and reporting Lp(a) in molar concentration rather than in mass units to facilitate standardization and harmonization in Lp(a) testing.13
Current and emerging therapies
Statins are one of the most potent treatments for the primary prevention of ASCVD through the reduction of LDL-C concentration. However, recent studies reveal that statins have no effect on Lp(a) concentration3 and others suggest that statin administration can increase Lp(a) concentration by up to 11%.5,9 Nonetheless, the EAS does not recommend statin therapy be halted as their strong ameliorative effects on CVD risk are well established and surmount the risk related to increased Lp(a) concentration.8
Niacin (nicotinic acid) is another established treatment for the reduction of CVD events and acts by increasing HDL levels. Niacin can reduce Lp(a) concentration though the reduction of gene expression in a dose-dependent manner.5 However, niacin therapy has not been proven to have beneficial effects on CVD risk.8
A recent metanalysis showed a 26% reduction in serum Lp(a) concentration through treatment with PCSK9 inhibitors. This is thought to be due to a shortage of apoB-100 molecules either because of reduced synthesis or competitive binding with other LDL receptors, resulting in reduced Lp(a) concentration.5 Several studies show the efficacy of PCSK9 inhibitors in reducing CVD risk, but this is not yet an approved therapy.5,8
New therapeutic strategies aim to target hepatocytes, the site of apo(a) synthesis, to reduce Lp(a) concentration. Antisense oligonucleotides (ASOs) inhibit apo(a) mRNA in the nucleus and cytoplasm, ultimately inhibiting Lp(a) secretion5 through the cleavage of the sense strand by ribonuclease H1.9 While still in clinical trials, ASO therapies show promise in the battle to reduce CVD risk with some studies displaying an overall reduction in Lp(a) concentration of more than 80%.9
Conclusion
There have been major advances in the understanding of Lp(a) pathophysiology in the last ten years establishing this macromolecular complex as an independent causal risk factor for various forms of CVD, however, more investigation is required to fully understand the mechanisms responsible for this association. With many national healthcare organizations and the EAS recommending universal testing for Lp(a) in adults, more emphasis should be placed on raising awareness of the importance of Lp(a) screening. Finally, more research is needed into therapies that succeed at lowering Lp(a) concentration. While some therapies are in clinical trials, there are currently no approved therapies that achieve this goal.
REFERENCES
1. Cardiovascular diseases (CVDs). Who.int. Published June 11, 2021. Accessed March 28, 2023. https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
2. Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. 2016;57(8):1339-59. doi:10.1194/jlr.R067314.
3. Zafrir B, Aker A, Saliba W. Extreme lipoprotein(a) in clinical practice: A cross sectional study. Int J Cardiol Cardiovasc Risk Prev. 2023;13;16:200173. doi:10.1016/j.ijcrp.2023.200173.
4. Pino BD, Gorini F, Gaggini M, Landi P, Pingitore A, Vassalle C. Lipoprotein(a), Cardiovascular Events and Sex Differences: A Single Cardiological Unit Experience. J Clin Med. 2023;18;12(3):764. doi:10.3390/jcm12030764.
5. Stürzebecher PE, Schorr JJ, Klebs SHG, Laufs U. Trends and consequences of lipoprotein(a) testing: Cross-sectional and longitudinal health insurance claims database analyses. Atherosclerosis. 2023;367:24-33. doi:10.1016/j.atherosclerosis.2023.01.014.
6. Lampsas S, Xenou M, Oikonomou E, et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules. 2023;28(3):969. doi:10.3390/molecules28030969.
7. Vuorio A, Watts GF, Schneider WJ, Tsimikas S, Kovanen PT. Familial hypercholesterolemia and elevated lipoprotein(a): double heritable risk and new therapeutic opportunities. J Intern Med. 2020;287(1):2-18. doi:10.1111/joim.12981.
8. Kronenberg F, Mora S, Stroes ESG, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J. 2022;43(39):3925-3946. doi:10.1093/eurheartj/ehac361
9. Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol. 2017;14;69(6):692-711. doi:10.1016/j.jacc.2016.11.042.
10. Boffa MB, Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57(5):745-57. doi:10.1194/jlr.R060582.
11. Enkhmaa B, Anuurad E, Berglund L. Lipoprotein (a): impact by ethnicity and environmental and medical conditions. J Lipid Res. 2016;57(7):1111-25. doi:10.1194/jlr.R051904.
12. Svilaas T, Klemsdal TO, Bogsrud MP, et al. High levels of lipoprotein(a) – assessment and treatment. Tidsskr Nor Laegeforen. 2022;16;142(1). English, Norwegian. doi:10.4045/tidsskr.21.0800.
13. Wyness SP, Genzen JR. Performance evaluation of five lipoprotein(a) immunoassays on the Roche cobas c501 chemistry analyzer. Pract Lab Med. 2021;24;25:e00218. doi:10.1016/j.plabm.2021.e00218.
About the Author

Jason Armstrong
BSc is a Scientific Content Creator at Randox Laboratories Ltd. After achieving his degree in Biochemistry and beginning his career in an R&D laboratory, he made the switch to pursue his passion in scientific communications.